2026-02-24 阅读量:393
癫痫是脑卒中后第二常见的神经系统疾病,中国每年新增确诊病例约240万。目前已有20余种抗癫痫药物应用于临床,但仍有约30%的患者癫痫发作无法得到有效控制。除外科手术外,可供选择的治疗手段十分有限。多数药物难治性癫痫患者并不适合接受外科干预。有数据显示,80%接受药物治疗的患者会出现不良反应,其中30%~40%的患者发生的不良反应会严重影响生活质量,进而导致停药或服药依从性下降,最终无法改善长期预后。此外,现有抗癫痫药物的作用机制与其临床疗效谱的关联性较差,且各类具有全新作用机制的抗癫痫药物的研发与临床应用,并未降低药物难治性癫痫的占比。因此,深入探究癫痫的发病机制、发掘新的治疗策略,仍是当前临床亟待解决的重点问题。
近期,安徽医科大学第一附属医院神经内科王玉教授,联合皖南医学院第一附属医院(皖南医学院弋矶山医院)神经内科周志明教授、麻醉科王斌副教授,在Advanced Science期刊(IF=14.1,中科院1区Top)上发表了题为“Microglial GPR35 Ameliorates Epileptogenesis and Neuroinflammation via PDGFA Domain 2 Signaling”的研究论文。研究发现,癫痫患者与动物模型的癫痫脑区中GPR35的表达均显著上调。进一步研究证实,GPR35可通过特定结合基序与PDGFA的第二结构域PDG2结合,抑制PDGFA经泛素—蛋白酶体途径的降解,并激活磷脂酰肌醇3-激酶-蛋白激酶B(PI3K-AKT)信号通路;该通路可有效减轻神经炎症反应,减少癫痫发作。此外,GPR35的表达缺失会破坏这条调控神经炎症与神经元过度兴奋的信号通路,而过表达PDGFA可模拟GPR35的激活效应,进而缓解炎症反应并抑制癫痫发生。上述研究结果明确GPR35为癫痫治疗的全新可成药靶点,为研发打破“神经炎症-神经元过度兴奋”恶性循环的抗癫痫治疗策略,提供了新的方向。安徽医科大学王琦博士、瞿婷婷硕士、孙其兵博士和李冉博士为论文共同第一作者,安徽医科大学第一附属医院王玉教授、皖南医学院第一附属医院(皖南医学院弋矶山医院)周志明教授和王斌副教授为论文共同通讯作者。该研究得到了国家自然科学基金、安徽省自然科学基金、安徽省临床转化专项、安徽省卫生健康委科研项目等多个项目的资助。
越来越多的研究证据表明,神经炎症过程在癫痫发生与癫痫发作中发挥着基础性作用。基础研究与临床研究均一致证实,白细胞介素(IL)-1β、肿瘤坏死因子(TNF)-α、IL-6等促炎介质的浓度升高,会通过破坏血脑屏障、诱导神经元过度兴奋、损伤突触可塑性等多种机制促进癫痫发生。团队前期研究也观察到,慢性癫痫患者的外周免疫细胞存在异常改变,且这些改变与癫痫发作复发呈显著相关性,这也凸显了慢性炎症在复发性癫痫发作活动中的作用。目前,靶向特定炎症细胞或分子通路的新型抗神经炎症药物已逐步研发,这类药物有望突破现有抗癫痫药物缺乏疾病修饰效应的局限性。由此可见,贯穿癫痫发生全过程的慢性神经炎症,是导致癫痫脑区神经元持续过度兴奋的关键因素,进一步开展相关机制研究与靶向干预探索,对于研发新型癫痫治疗策略具有至关重要的意义。
G蛋白偶联受体35(GPR35)是一种可被内源性犬尿氨酸(KYNA)激活的GPCR,参与调控炎症反应。既往研究显示,在CX3CR1阳性巨噬细胞条件性敲除GPR35的小鼠模型中,小鼠病理状态下的炎症反应显著加剧,这提示GPR35在髓系细胞介导的免疫反应中发挥重要调控作用。在脂多糖(LPS)诱导的小胶质细胞炎症模型中,激活GPR35可抑制NLRP3炎症小体的组装,减少IL-1β和IL-18的分泌,并促进抗炎因子IL-10的产生。在实验性自身免疫性脑脊髓炎小鼠模型中,敲除GPR35会加重辅助性T细胞17亚群(Th17细胞)的浸润与血脑屏障的破坏,进一步证实了其抗神经炎症的潜在作用。GPR35的调控功能失调还与阿尔茨海默病、帕金森病等神经退行性疾病密切相关。然而,GPR35在癫痫中的表达模式及作用机制仍未明确。
血小板衍生生长因子A(PDGFA)是一种经典的与发育和血管生成相关的因子。PDGF家族已被证实是神经炎症的关键调控因子,其发挥的作用具有环境依赖性,既可加重病理损伤,也可缓解病理进程。PDGFA主要通过其受体血小板衍生生长因子受体α(PDGFRα)传递信号,对少突胶质前体细胞的稳态维持与血管生成至关重要。在神经炎症微环境中,PDGFA的表达会显著上调,且星形胶质细胞来源的PDGFA可强效诱导小胶质细胞活化,参与启动神经炎症级联反应。除发挥免疫调节作用外,PDGFA信号通路还参与中枢神经系统稳态的维持,该通路的调控失调与神经炎症的发病机制密切相关。但PDGFA与GPCR信号通路的相互作用程度,尤其是其在癫痫中与小胶质细胞GPR35的功能性相互作用机制,目前仍不清楚。
1.颞叶癫痫患者及癫痫体内、体外模型中GPR35的表达
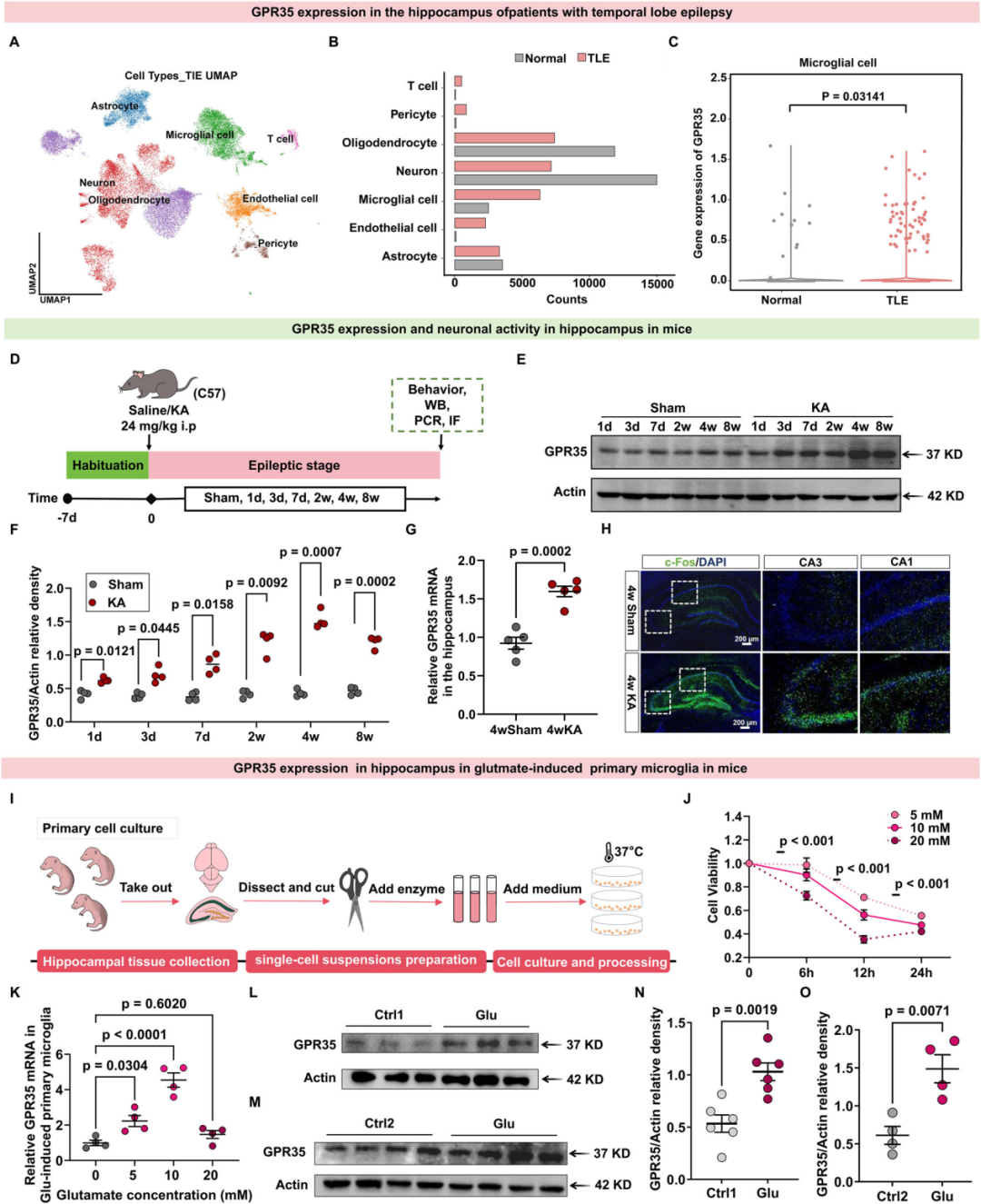
神经炎症是癫痫发作的常见发病机制之一。已有研究证实,GPR35可通过其内源性配体KYNA调控组织能量稳态与炎症反应,而KYNA在癫痫相关炎症中发挥关键作用。据此推测,GPR35是参与癫痫发生过程中神经炎症反应的关键调控蛋白。为明确GPR35与癫痫发作的相关性,研究者首先利用公共人类单核RNA测序(snRNA-seq)数据集,分析该受体在颞叶癫痫(TLE)患者海马组织中的分布特征。通过代表性标记基因聚类分析,研究人员鉴定出9种主要细胞类型。细胞群分析结果显示,TLE患者与非癫痫对照组的细胞构成存在显著差异,其中TLE患者出现神经元丢失,且小胶质细胞数量增多。进一步分析发现,GPR35在TLE患者的小胶质细胞中呈显著上调表达,而在其他细胞类型中无此变化。
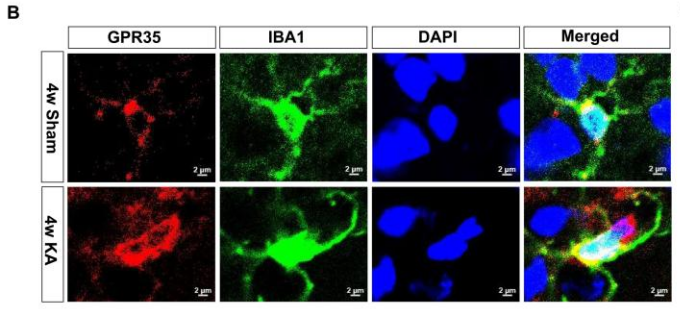
随后,作者探究了癫痫模型中GPR35的表达变化。采用海人酸(KA)诱导法建立小鼠癫痫模型,假手术组小鼠同期予腹腔注射等体积生理盐水。采用拉辛评分量表评估急性癫痫发作期的发作程度,仅将出现Ⅳ~Ⅵ级癫痫发作的小鼠纳入后续分析。于KA诱导及假手术处理后3 d、1 w、2 w、4 w、8 w这5个时间节点,分别检测各组小鼠的GPR35表达水平。蛋白质免疫印迹实验(Western blot,WB)结果显示,小鼠海马组织中GPR35的表达量在KA诱导后4 w达到峰值,至8 w时有所下降。KA诱导后4 w,与假手术组相比,癫痫模型小鼠的促炎细胞因子IL-1β、IL-6、TNF-α表达上调,而抗炎细胞因子IL-10表达下调。同时,模型小鼠海马组织中GPR35的mRNA表达量亦显著升高,且该受体的表达量与促炎因子mRNA表达量呈正相关,与抗炎因子mRNA表达量呈负相关。与snRNA-seq结果一致,海马组织免疫荧光染色结果显示,KA诱导的癫痫模型小鼠小胶质细胞中GPR35的蛋白表达水平显著高于假手术组,而神经元中无明显差异。且GPR35激活与神经元特异性活化标志物c-Fos的表达上调呈同步发生。
研究者进一步构建谷氨酸诱导的细胞模型,对GPR35蛋白的表达动态进行时间依赖性分析。采用细胞计数试剂盒-8(CCK-8)检测不同浓度谷氨酸[5、10、20 mmol/L(mM)]对原代小胶质细胞[分离自出生0天(P0)C57BL/6小鼠的海马组织]活力的时间依赖性影响,结果显示,谷氨酸对小胶质细胞活力的抑制作用呈剂量和时间依赖性;作用6 h后细胞活力即显著下降,20 mM高浓度谷氨酸作用24 h可导致细胞活力下降约60%。聚合酶链式反应(PCR)检测结果显示,10 mM谷氨酸诱导12 h后,细胞中GPR35的mRNA表达量达到峰值,且与促炎因子IL-1β、TNF-α的表达上调趋势一致。因此,作者选取10 mM谷氨酸诱导12 h的细胞模型开展后续实验。WB结果显示,与对照组相比,BV2小胶质细胞系模型中GPR35的蛋白表达水平显著升高;原代小胶质细胞模型中亦得到相似结果。以上结果表明,在人类、小鼠体内模型及谷氨酸诱导的体外细胞模型中,GPR35的表达上调均与癫痫发作存在相关性。
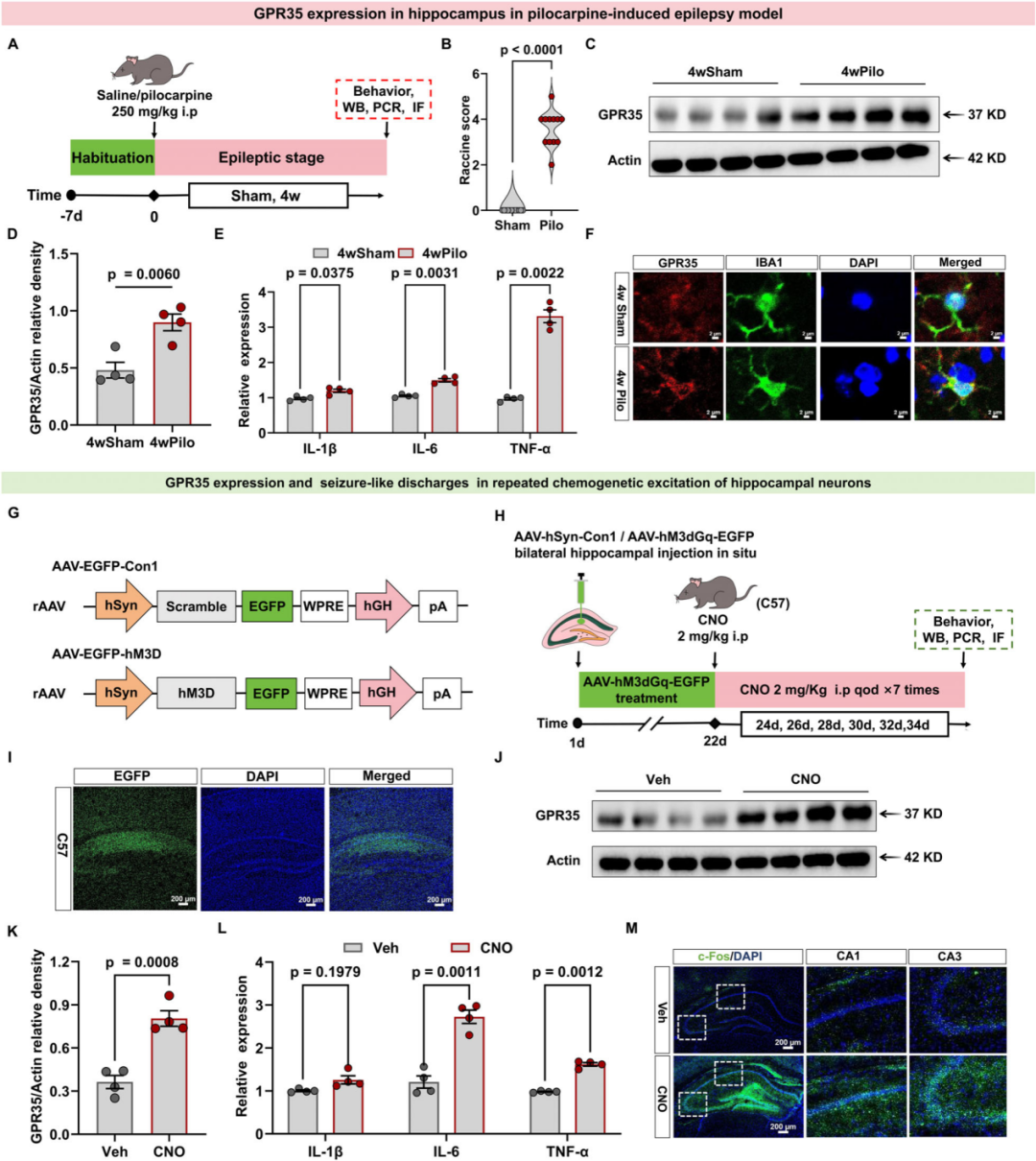
研究人员还验证了KA诱导癫痫模型中GPR35的表达变化是否可在其他癫痫模型中重现。结果与预期一致,毛果芸香碱(Pilo)诱导的癫痫模型中该受体呈高表达;对海马神经元进行反复化学遗传学激活后,同样观察到GPR35的高表达。上述结果提示,癫痫发作后海马组织中GPR35的表达呈时间依赖性上调,且该变化与癫痫发作的诱导方式无关。
图1.GPR35在癫痫患者、KA诱导的体内癫痫模型及谷氨酸诱导的体外癫痫模型中的表达
图2.KA诱导癫痫小鼠海马组织小胶质细胞中GPR35表达上调
图3.GPR35在其他癫痫模型中的表达
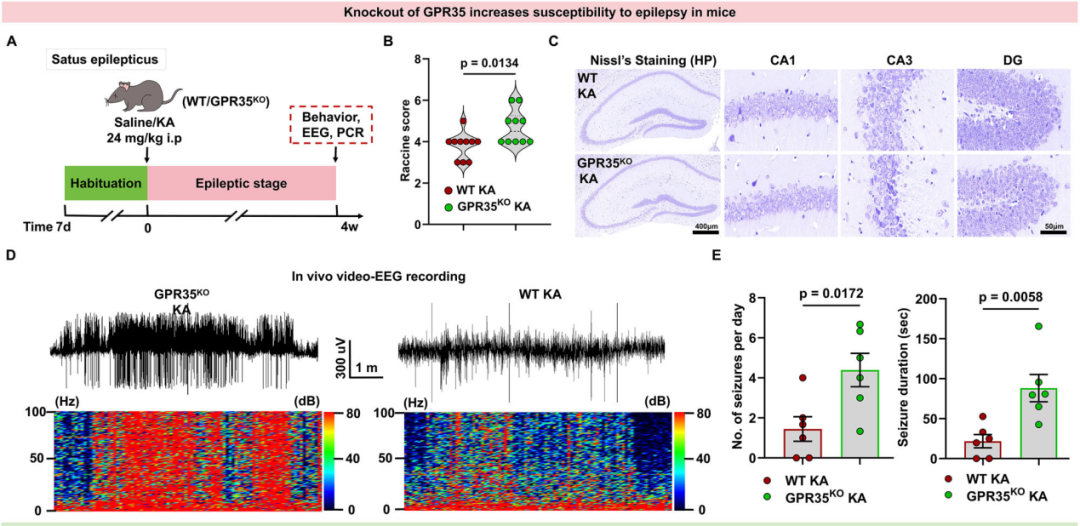
2.GPR35基因敲除加重癫痫小鼠慢性癫痫发作严重程度
为明确GPR35在KA诱导4 w的癫痫小鼠海马组织中是否发挥功能性作用,研究人员构建了GPR35全身基因敲除(GPR35KO)小鼠,以验证该基因在癫痫发生过程中的作用。首先,实验证明该基因敲除对小鼠的体质量及脑形态均无显著影响。为验证GPR35基因缺失是否影响癫痫发作的严重程度,分别对GPR35KO小鼠和野生型(WT)小鼠构建KA诱导癫痫模型。结果显示,与KA诱导的WT小鼠相比,KA诱导的GPR35KO小鼠拉辛评分更高,神经元丢失现象更显著,促炎因子表达水平也更高。皮层脑电图(EEG)记录结果显示,KA诱导4 w后,GPR35KO小鼠的癫痫发作严重程度较WT小鼠显著增加(每日癫痫发作次数显著增加,癫痫发作持续时间显著延长)。上述结果表明,在KA诱导4 w的癫痫模型中,GPR35参与调控癫痫相关的神经炎症病理生理过程。综合以上研究结果可见,GPR35基因缺失加重癫痫小鼠慢性癫痫发作的严重程度,提示该基因参与癫痫的发病机制。
图4.GPR35表达缺失加重小鼠癫痫发作
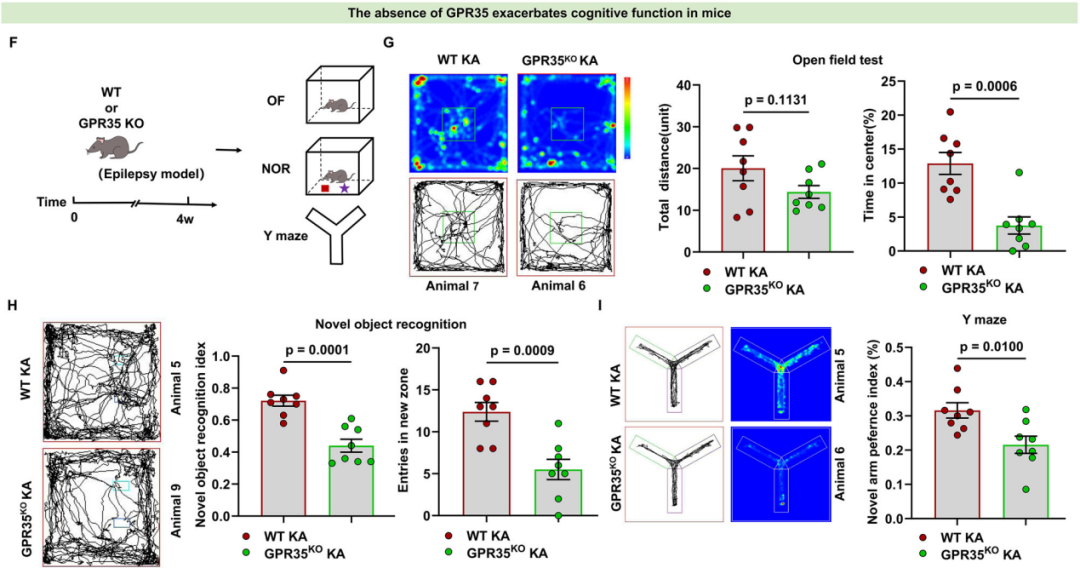
3.GPR35缺失加剧KA诱导癫痫小鼠的认知损伤
接下来,研究人员进一步探究了GPR35缺失对癫痫小鼠认知功能的影响。为明确GPR35调控癫痫相关认知功能的作用机制,分别对WT小鼠与GPR35KO小鼠构建KA诱导的癫痫模型,随后开展一系列行为学实验。采用新物体识别实验(NOR)和Y迷宫实验评估小鼠的海马依赖性学习与记忆能力,同时通过旷场实验(OFT)检测小鼠的焦虑样行为。结果显示,与癫痫对照组小鼠相比,GPR35KO癫痫小鼠对中央区域的探索意愿显著降低,在中央区域停留时间占比降低,而两组小鼠的总运动距离无明显差异(自由探索10 min)。小鼠在旷场中央区域停留时间缩短,是其焦虑样更显著的行为学特征,这提示GPR35缺失会加重癫痫小鼠的焦虑样行为。NOR期间,小鼠可在实验箱内对新物体和熟悉物体自由探索10 min;Y迷宫实验中,小鼠从起始臂向另外两个臂自由活动10 min。行为学检测结果显示,与癫痫对照组小鼠相比,GPR35KO癫痫小鼠对新物体和新臂的偏好度均显著低于对熟悉物体和熟悉臂的偏好度。上述结果表明,GPR35KO癫痫小鼠存在空间工作记忆损伤,且GPR35缺失会加重癫痫小鼠的空间记忆损伤。综合以上结果可见,GPR35基因敲除会加剧KA诱导癫痫小鼠的认知功能损伤,具体表现为小鼠的探索行为减少、物体识别能力下降及空间记忆受损,提示GPR35在癫痫相关行为学缺陷中发挥神经保护作用。
图5.GPR35表达缺失损害癫痫小鼠认知功能
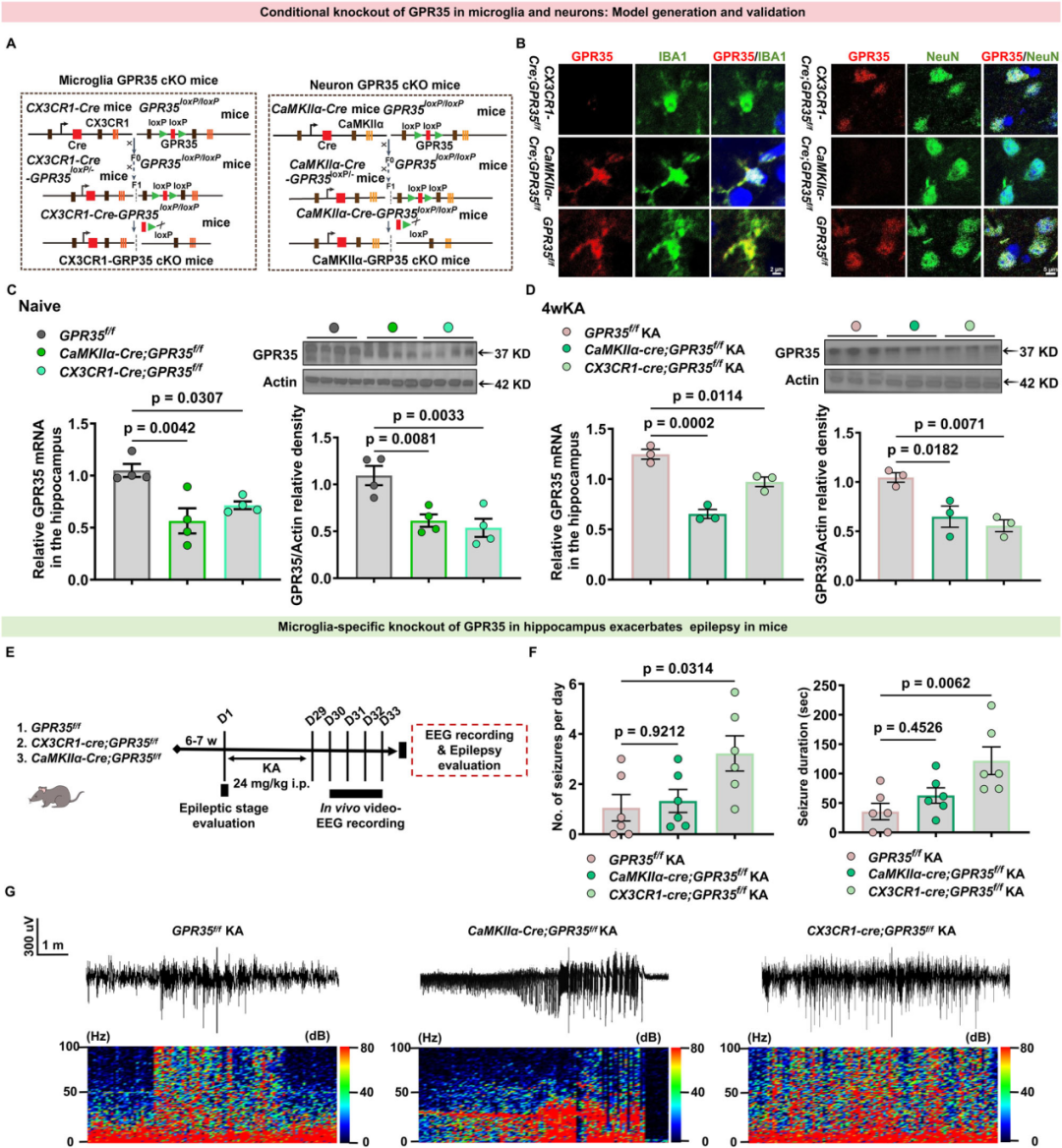
4.GPR35对癫痫易感性的调控作用具有细胞类型特异性
为探究GPR35的细胞特异性缺失是否影响癫痫发作严重程度,研究者以C57BL/6J小鼠为背景,构建了两种条件性基因敲除小鼠模型:①小胶质细胞特异性敲除模型(CX3CR1-Cre;GPR35f/f)和②海马锥体神经元特异性敲除模型(CaMKIIα-Cre;GPR35f/f)。选取同窝、同周龄、同性别且无Cre重组酶的GPR35f/f小鼠作为对照组。实验前,所有小鼠均在标准化无特定病原体(SPF)环境中进行6 w的适应性饲养。借助上述两种模型,可精准判定GPR35缺失的致癫痫调控效应是否由海马锥体神经元、小胶质细胞单独介导,或由二者共同介导。
研究人员首先通过免疫荧光双标实验验证小胶质细胞中GPR35的敲除效率。联合使用GPR35抗体与小胶质细胞标志物IBA1抗体进行染色,结果显示,CX3CR1-Cre;GPR35f/f小鼠的小胶质细胞中GPR35完全缺失,而神经元中仍可检测到GPR35表达;与之相反,CaMKIIα-Cre;GPR35f/f小鼠的小胶质细胞仍保留较强的GPR35免疫反应性。对CaMKIIα-Cre;GPR35f/f小鼠海马CA1区的检测结果证实,其神经元中GPR35已实现高效敲除,而小胶质细胞中GPR35的表达不受影响。值得注意的是,荧光定量PCR(qPCR)和WB检测结果显示,在基础状态及癫痫持续状态(SE)后4 w,CX3CR1-Cre;GPR35f/f小鼠和CaMKIIα-Cre;GPR35f/f小鼠的海马组织总GPR35 mRNA和蛋白表达水平均下降约20%~40%,该结果与预期的细胞特异性敲除效应一致。
为明确小胶质细胞或神经元中的GPR35表达是否参与癫痫发生,作者对CaMKIIα-Cre;GPR35f/f小鼠、CX3CR1-Cre;GPR35f/f小鼠及GPR35f/f对照小鼠均进行KA诱导造模。采用拉辛评分量表对小鼠急性期的行为学癫痫发作程度进行评估,并通过在体EEG记录分析小鼠慢性期(KA诱导后4 w)的脑电癫痫发作活动。皮层EEG结果显示,与GPR35f/f对照组相比,CX3CR1-Cre;GPR35f/f小鼠的癫痫发作活动显著加剧,而CaMKIIα-Cre;GPR35f/f小鼠仅表现出自发性癫痫发作频率升高的轻微趋势。具体而言,CX3CR1-Cre;GPR35f/f小鼠的每日癫痫发作频率显著增加,且单次发作持续时间也明显延长。上述结果表明,神经元特异性GPR35缺失仅使小鼠癫痫易感性呈轻度升高趋势,而小胶质细胞特异性GPR35缺失则可诱发更为严重的癫痫表型。由此可见,GPR35敲除对癫痫发生的调控效应具有细胞类型依赖性,且以小胶质细胞特异性敲除的作用效应最为突出。综合来看,小胶质细胞中GPR35的缺失会显著升高小鼠的癫痫易感性,而神经元中GPR35的缺失仅轻微降低其癫痫发作阈值。以上研究结果提示,癫痫发生过程中GPR35主要通过小胶质细胞发挥调控作用。
图6.GPR35对癫痫发作的细胞特异性调控作用
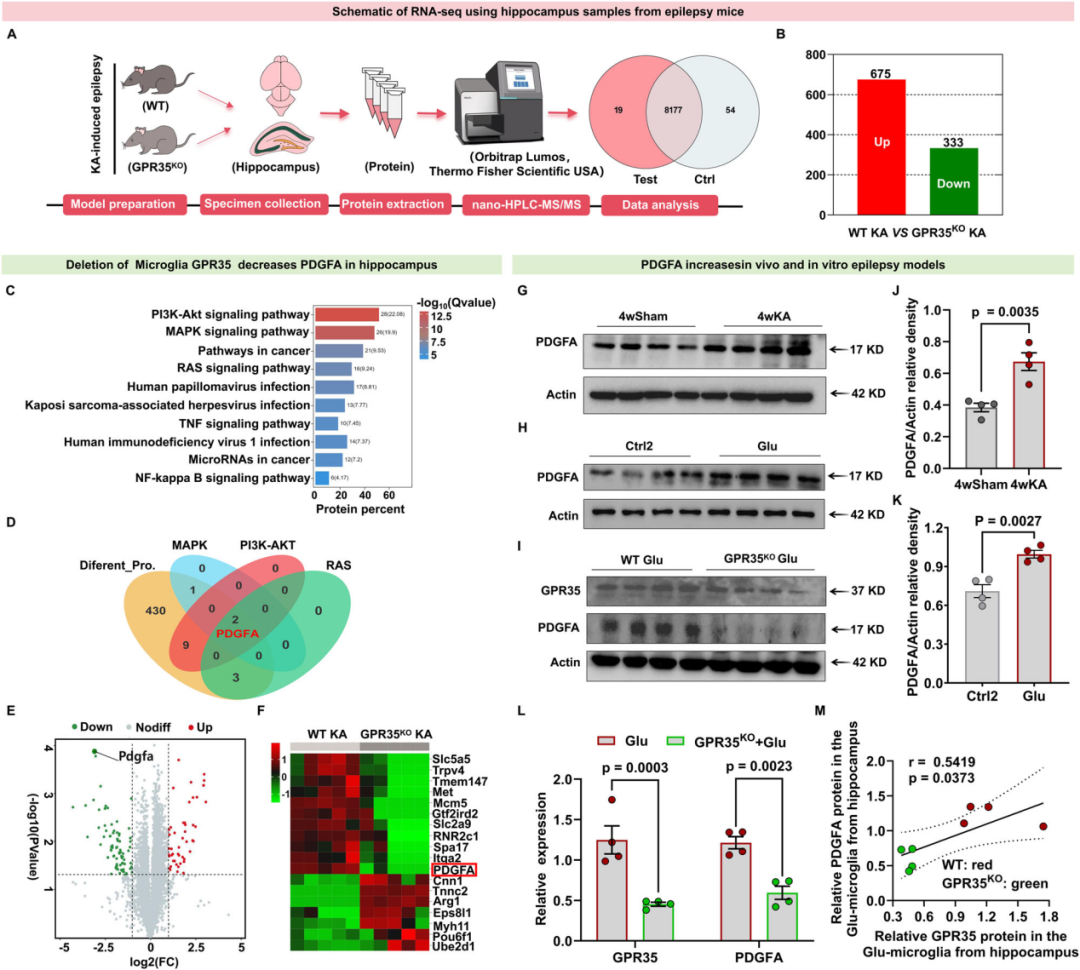
5.血小板衍生生长因子A是海马组织中GPR35的下游靶点,参与调控KA诱导4 w后的癫痫发作
为阐明海马组织中GPR35介导癫痫调控作用的潜在机制,研究人员对KA诱导的GPR35KO癫痫小鼠及WT癫痫小鼠的海马组织开展定量蛋白质组学分析。通过蛋白质组谱共鉴定出1508个可定量蛋白,以差异倍数≥1.5、p<0.05且多重检验校正后的错误发现率(FDR)<0.05作为差异表达蛋白的筛选标准。
图7.在KA诱导的体内癫痫模型及谷氨酸诱导的体外癫痫模型中,PDGFA均为GPR35的下游靶点
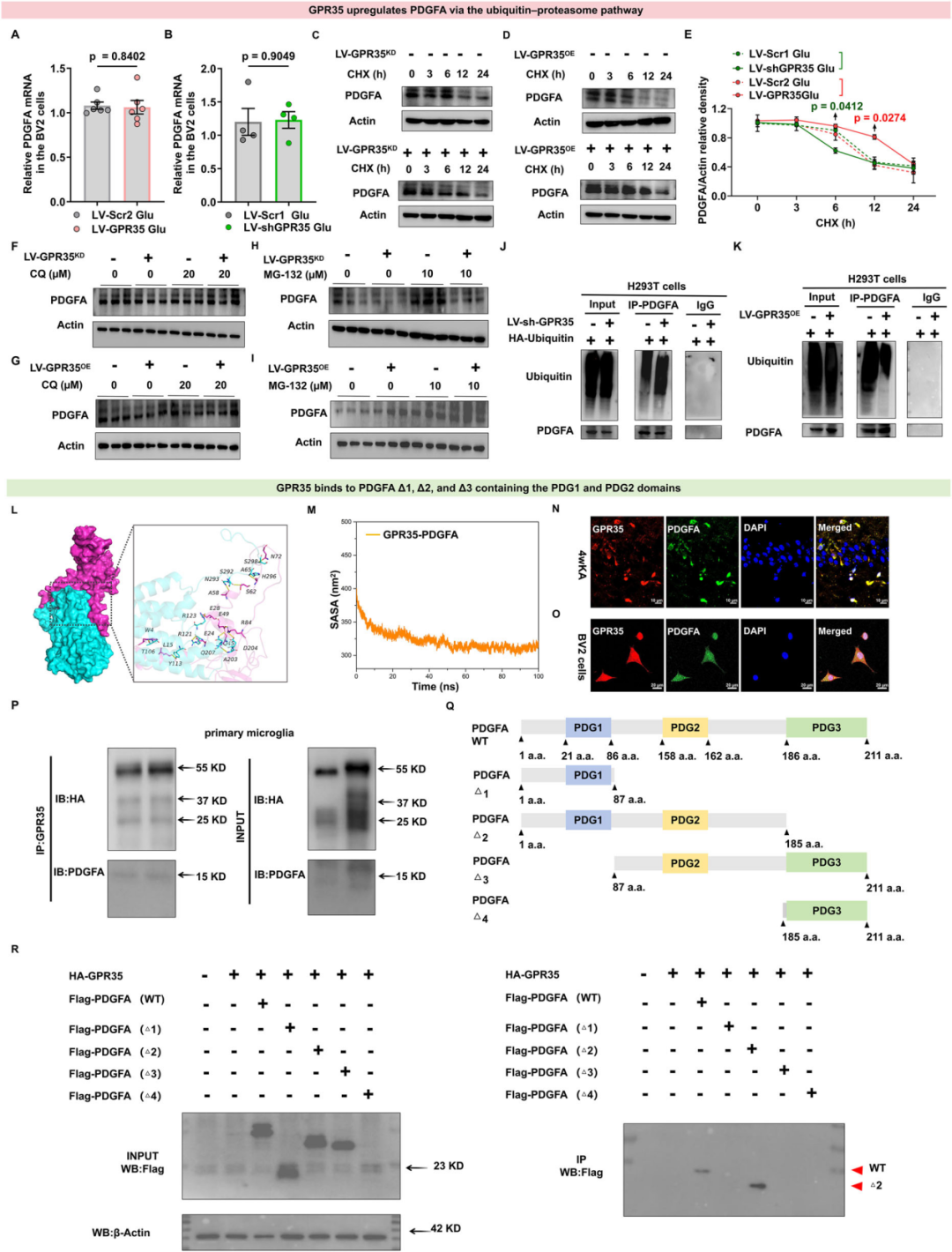
6.GPR35通过泛素—蛋白酶体通路抑制PDGFA的降解
为探究GPR35调控PDGFA表达的分子机制,研究人员对PDGFA的mRNA水平进行了定量检测。在谷氨酸诱导的BV2细胞中感染过表达或敲低GPR35的慢病毒载体,结果显示GPR35表达水平的改变未影响PDGFA的转录水平,提示GPR35可能调控PDGFA的蛋白稳定性。随后,研究者使用蛋白合成抑制剂放线菌酮(CHX,200 μg/mL)处理谷氨酸诱导的BV2细胞,分别作用3、6、12和24 h,以溶媒处理的细胞作为对照组。药物处理后收集细胞并裂解,通过WB检测PDGFA的蛋白表达水平。结果显示,BV2细胞中敲低GPR35(GPR35KD)后,PDGFA的蛋白半衰期显著缩短;反之,过表达GPR35(GPR35OE)的细胞经CHX处理后,PDGFA的蛋白半衰期显著延长。
接下来,研究人员在谷氨酸诱导的原代小胶质细胞中,进一步探究GPR35调控PDGFA表达的分子机制,对细胞内两条主要的蛋白降解通路—蛋白酶体降解通路和自噬降解通路进行了检测分析。结果显示,自噬抑制剂氯喹(CQ,20 μM)对GPR35调控的PDGFA表达无显著影响;而26S蛋白酶体抑制剂MG-132(10 μM)处理原代小胶质细胞后,可阻断由GPR35KD引发的PDGFA降解增强效应,且MG-132可进一步上调GPR35OE细胞中PDGFA的表达水平。
基于上述结果可得出结论:GPR35通过泛素—蛋白酶体通路维持PDGFA的蛋白稳定性。最后,作者对转染了血凝素(HA)标签泛素质粒的细胞进行内源性PDGFA免疫共沉淀(Co-IP)实验,结果显示,GPR35KD的人胚肾293T(HEK293T)细胞中,PDGFA的泛素化修饰水平显著升高;与之相符,GPR35OE的HEK293T细胞中,PDGFA的泛素化修饰水平较对照组显著降低。以上结果表明,GPR35通过抑制PDGFA的泛素—蛋白酶体途径降解,维持其蛋白表达的稳定性。此外,从分子机制上进一步证实,GPR35通过直接结合PDGFA的PDG2结构域,激活其下游信号通路。
图8.GPR35通过结合PDGFA的PDGF2结构域,经泛素—蛋白酶体通路抑制PDGFA的降解
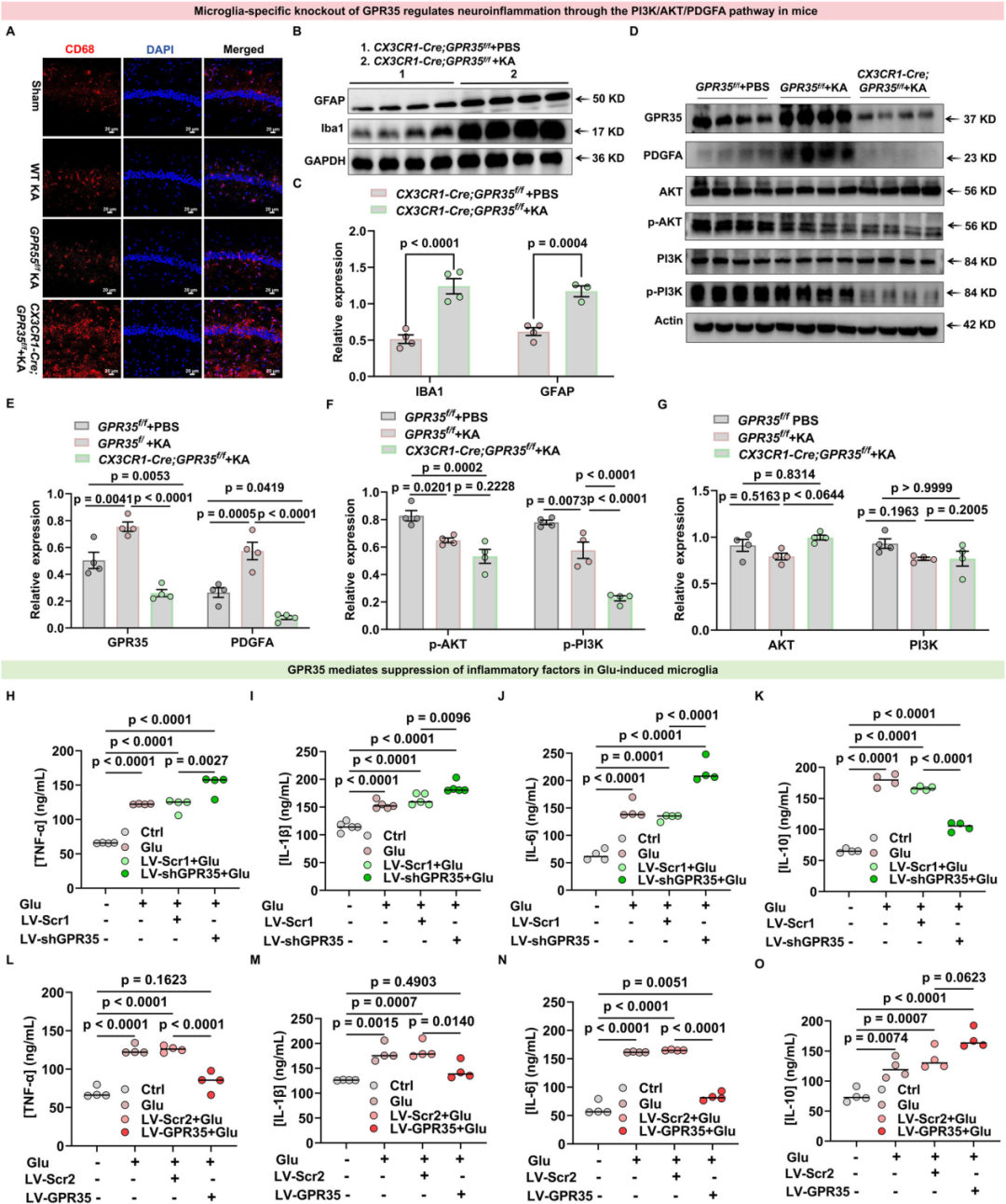
7.小胶质细胞特异性GPR35敲除通过PI3K-AKT信号通路加重癫痫发作与神经炎症
为探究癫痫小鼠模型中GPR35对脑内神经炎症的调控作用,作者首先证实Cre表达不会产生混杂效应:CX3CR1-Cre小鼠的全身GPR35表达水平及基础炎症相关蛋白表达量,与GPR35f/f同窝对照小鼠无显著差异。排除Cre的干扰效应后,研究人员检测了小胶质细胞溶酶体标志物CD68阳性细胞的数量。免疫荧光结果显示,KA注射后,CX3CR1-Cre;GPR35f/f小鼠海马组织中的CD68阳性细胞数量显著增多,提示小胶质细胞的吞噬活性增强。
KA造模4 w后对小鼠脑组织的WB检测结果显示,与GPR35f/f小鼠相比,CX3CR1-Cre;GPR35f/f小鼠的小胶质细胞标志物IBA1、星形胶质细胞标志物胶质纤维酸性蛋白(GFAP)表达均上调,其中IBA1的上调尤为显著。基于前述KEGG通路分析结果,PI3K-AKT信号通路被鉴定为癫痫模型中富集程度最显著的炎症相关通路。后续生物信息学分析筛选PDGFA的潜在下游底物,进一步确定PI3K和AKT为关键效应分子。为此,研究人员检测了PI3K和AKT的磷酸化水平。结果显示,与PBS处理的GPR35f/f小鼠相比,KA造模4 w的GPR35f/f小鼠海马组织中,PI3K丝氨酸(Ser)467/199及AKT Ser473位点磷酸化水平显著降低,同时伴随GPR35与PDGFA蛋白表达显著上调;而KA诱导的CX3CR1-Cre;GPR35f/f小鼠中,上述位点的磷酸化水平进一步降低,且GPR35与PDGFA蛋白表达亦下调。这些结果表明,GPR35、PDGFA、p-PI3K(Ser467/199)与p-AKT(Ser473)的表达水平呈显著正相关。以上数据提示,小胶质细胞中GPR35的缺失会导致PDGFA表达异常,进而使PI3K(Ser467/199)和AKT(Ser473)的磷酸化水平降低,最终抑制抗炎细胞因子的释放、促进促炎细胞因子的产生;这种调控紊乱会加剧脑内神经炎症反应,进而促进癫痫发生。
研究者进一步利用谷氨酸刺激的原代小胶质细胞模型,验证GPR35对炎症反应的调控作用。酶联免疫吸附试验(ELISA)结果显示,谷氨酸刺激条件下,通过短发夹RNA(shRNA)敲低GPR35的表达,显著增加TNF-α、IL-1β、IL-6的分泌量,而IL-10的分泌量则显著降低。反之,谷氨酸刺激后过表达GPR35,可显著减少TNF-α、IL-1β、IL-6的分泌,并上调IL-10的分泌水平。上述结果表明,癫痫样病理条件下,小胶质细胞中GPR35的激活可双向调控关键炎症细胞因子的表达。
图9.小胶质细胞特异性敲除GPR35通过PI3K-AKT-PDGFA信号通路调控小鼠的癫痫发生与神经炎症
8.小胶质细胞特异性过表达PDGFA可激活海马区PDGFA-PI3K-AKT信号级联反应
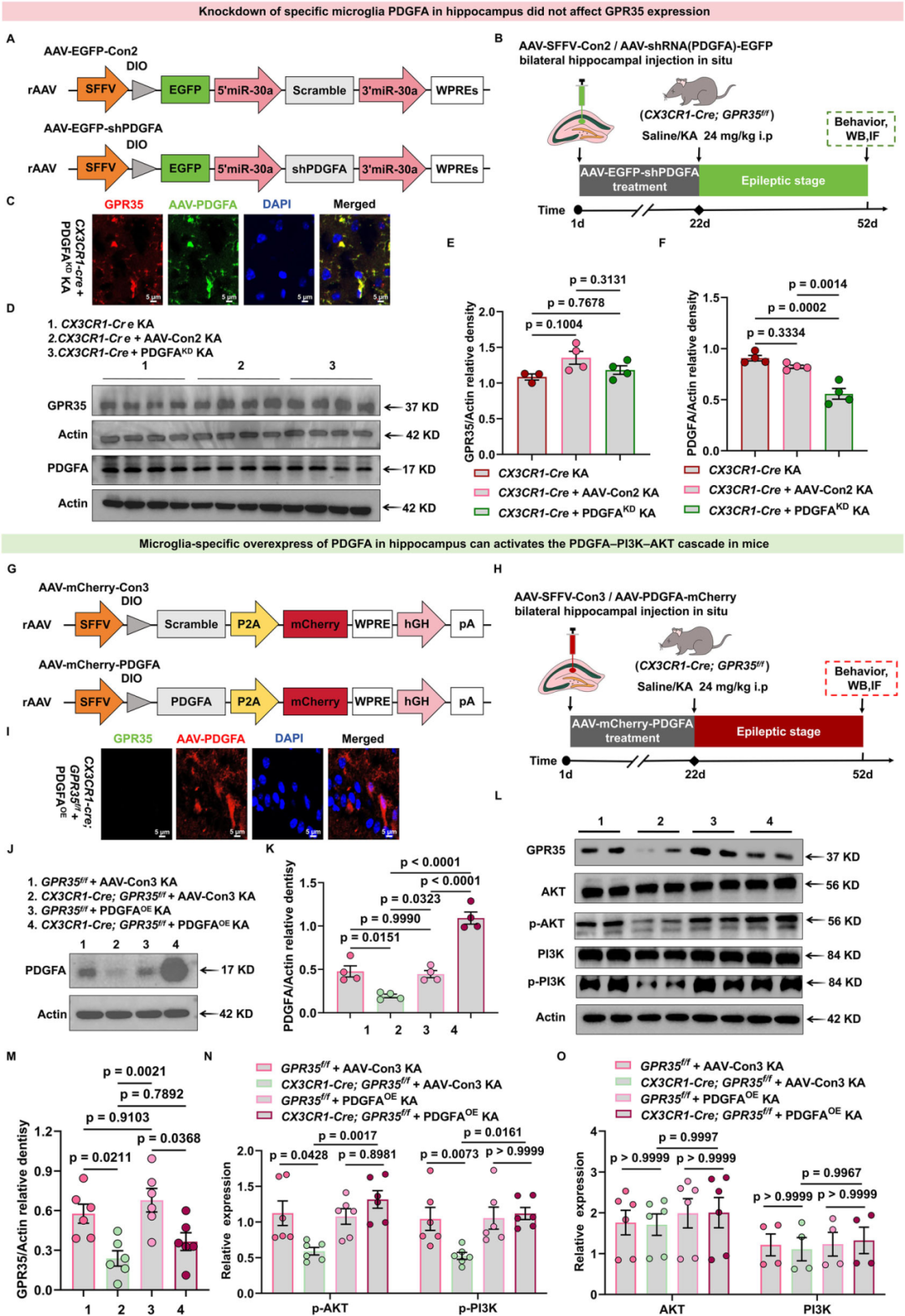
GPR35通过与不同的配体受体复合物发生结构性偶联实现信号转导,进而触发下游信号级联反应。作者首次证实,PDGFA在GPR35介导的炎症相关通路中发挥主导作用。于是,研究人员将PDGFA作为GPR35依赖性癫痫调控效应的潜在介导因子展开研究。鉴于癫痫病理状态下PDGFA的表达水平会升高,进一步探究PDGFA缺失是否会改变GPR35激活所产生的功能性效应。采用立体定位注射技术,向CX3CR1-Cre小鼠双侧海马区分别输注重组腺相关病毒rAAV-SFFV-DIO-EGFP(空载对照病毒,AAV-Con2)与rAAV-SFFV-DIO-shPDGFA-EGFP(PDGFA敲低病毒,PDGFAKD),以实现Cre依赖型的小胶质细胞特异性PDGFA敲低。病毒感染21天后构建KA诱导的癫痫发作模型,后续继续饲养小鼠至造模后共4 w。WB检测结果显示,与注射AAV-Con2的KA诱导CX3CR1-Cre小鼠相比,注射PDGFAKD的KA诱导CX3CR1-Cre小鼠中PDGFA蛋白表达水平显著降低,而GPR35蛋白表达无显著改变。上述结果表明,GPR35的表达不受PDGFA调控的影响,证实GPR35位于PDGFA的上游。
为恢复小胶质细胞特异性PDGFA表达,对CX3CR1-Cre;GPR35f/f小鼠及同窝对照小鼠双侧海马区进行立体定位注射,分别给予rAAV-SFFV-DIO-mCherry(空载病毒对照,AAV-Con3)与rAAV-SFFV-DIO-PDGFA-mCherry(PDGFA过表达病毒,PDGFAOE)。随后对小鼠进行KA诱导造模,后续继续饲养小鼠至造模后共4 w。WB检测证实,与注射AAV-Con3的KA诱导CX3CR1-Cre;GPR35f/f小鼠相比,注射PDGFAOE的KA诱导CX3CR1-Cre;GPR35f/f小鼠中PDGFA蛋白表达水平显著上调。随后检测了PI3K-AKT信号通路中的关键效应分子。结果显示,相比于注射AAV-Con3的KA诱导CX3CR1-Cre;GPR35f/f小鼠,注射PDGFAOE的KA诱导CX3CR1-Cre;GPR35f/f小鼠海马组织中p-PI3K(Ser467/199)及p-AKT(Ser473)水平显著升高。上述研究结果表明,GPR35可通过激活小胶质细胞内PDGFA-PI3K-AKT信号级联反应,发挥其生物学效应。
图10.抑制小胶质细胞中的PDGFA可阻断海马区PDGFA-PI3K-AKT信号级联反应
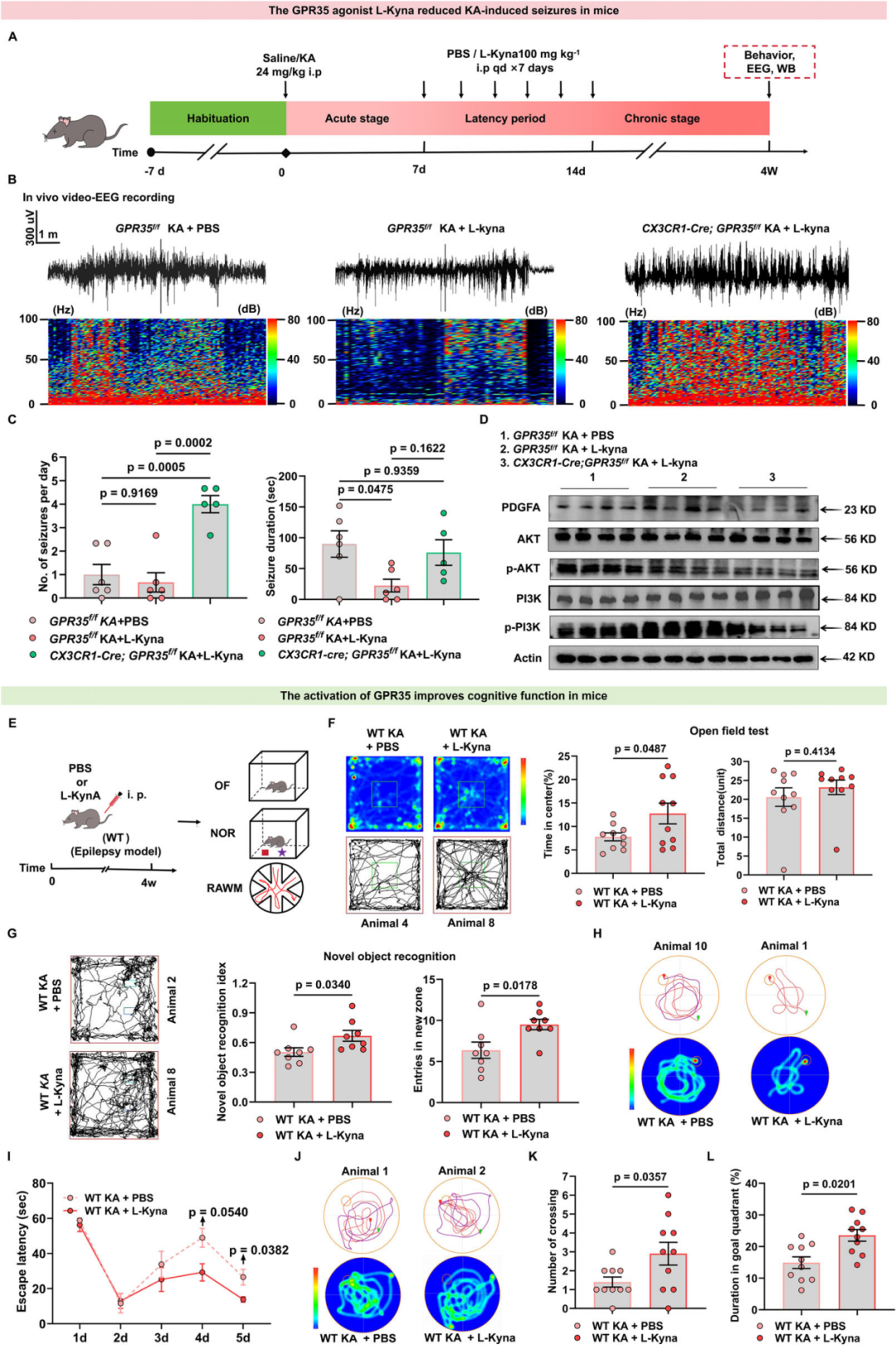
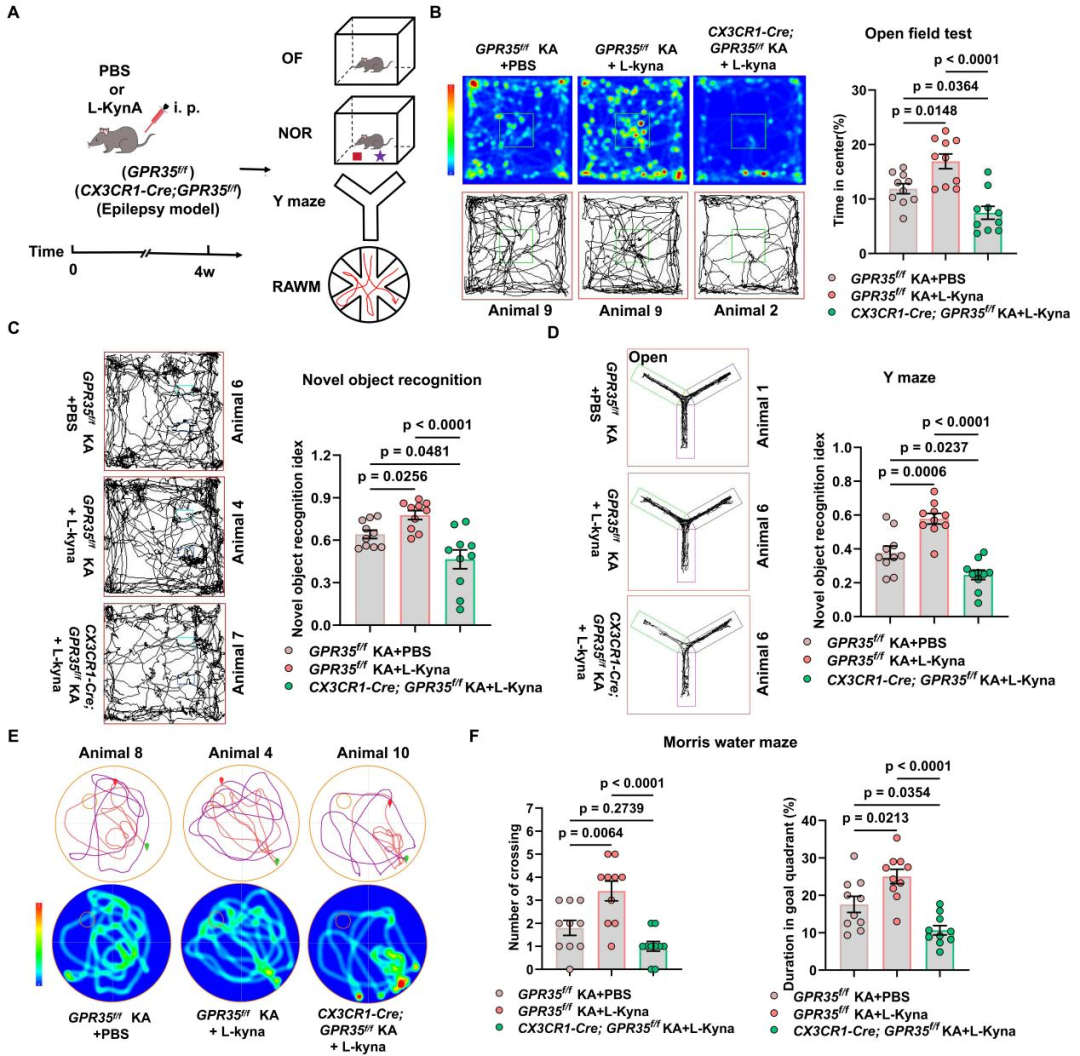
9.L-犬尿氨酸可抑制小鼠癫痫发作,并改善癫痫相关认知功能损伤
KYNA作为孤儿受体GPR35的内源性激动剂,在癫痫中具有重要的临床意义。研究表明,癫痫性痉挛动物模型的脑组织中,KYNA水平显著降低;癫痫患者脑脊液和血清样本中的KYNA浓度下降。值得注意的是,其前体物质L-犬尿氨酸(L-Kyn)可轻易穿过血脑屏障。于是,研究人员探究了补充L-Kyn是否能够抑制KA诱导的癫痫发作。GPR35f/f小鼠和CX3CR1-Cre;GPR35f/f小鼠经7天适应后,于第0天给予KA诱导癫痫急性期发作;自第7天(潜伏期起始)至第14天,每日腹腔注射L-Kyn(100 mg/kg)或PBS,持续7天,覆盖整个潜伏期;于KA造模后4 w(慢性期)通过行为学、EEG及WB检测,评价L-Kyn对KA诱导癫痫及相关损伤的干预作用。结果显示,与注射PBS的KA诱导GPR35f/f小鼠相比,注射L-Kyn的KA诱导GPR35f/f小鼠癫痫发作持续时间显著缩短,但第29-31天自发性反复癫痫发作(SRS)的频率无明显改变,提示L-Kyn可减轻KA诱导的慢性癫痫发作严重程度。WB结果显示,相比于注射PBS的KA诱导GPR35f/f小鼠,注射L-Kyn的KA诱导GPR35f/f小鼠海马组织中PDGFA蛋白水平升高,PI3K(Ser467/199)及AKT(Ser473)磷酸化水平显著上升;而在注射L-Kyn的KA诱导CX3CR1-Cre;GPR35f/f小鼠中,上述效应消失。此外,免疫荧光定量结果显示,注射L-Kyn的KA诱导GPR35f/f小鼠CD68阳性小胶质细胞数量明显减少,提示小胶质细胞吞噬活性降低。以上结果表明,L-Kyn通过GPR35依赖性激活PDGFA-PI3K-AKT通路并调控小胶质细胞活性,从而减轻癫痫发作严重程度。
接下来,作者在谷氨酸诱导的细胞模型中探究L-Kyn的作用。通过检测不同浓度L-Kyn(12.5、25、50 μM)对PDGFA及炎症因子mRNA表达的剂量依赖性效应,发现BV2细胞中PDGFA mRNA水平在25 μM L-Kyn处理后达到峰值。与溶剂组相比,25 μM L-Kyn处理的BV2细胞促炎细胞因子(IL-6、IL-1β、TNF-α)表达显著下调,同时抗炎细胞因子IL-10表达显著升高。因此选择25 μM L-Kyn用于后续实验。此外,WB证实,相比于溶剂组,L-Kyn处理可显著升高谷氨酸诱导的原代小胶质细胞中PDGFA蛋白水平,提示L-Kyn水平升高在谷氨酸诱导的体外模型中发挥保护作用。
进一步地,研究人员评估GPR35激活能否改善癫痫相关认知功能。对KA诱导WT小鼠在潜伏期分别注射L-Kyn或PBS。KA诱导后4 w,OFT结果显示,两组小鼠运动总距离无显著差异;但L-Kyn处理的WT癫痫小鼠在旷场中央区域停留时间占比显著升高,提示焦虑样行为减轻。NOR中L-Kyn处理的WT癫痫小鼠对新物体表现出明显偏好,而PBS处理的对照组无明显偏好。通过Morris水迷宫实验(MWM)评估学习与空间记忆能力,连续5天进行习得训练(定位航行实验),第6天进行探查实验(空间探索实验)。与PBS处理的WT癫痫小鼠相比,L-Kyn处理组逃避潜伏期显著缩短,提示学习能力改善。探索实验中,L-Kyn处理的癫痫小鼠穿越原平台位置次数更多,在目标象限停留时间占比更高,表明L-Kyn可改善癫痫小鼠的空间记忆损伤。上述行为学结果表明,L-Kyn可通过激活GPR35改善癫痫小鼠的认知功能。
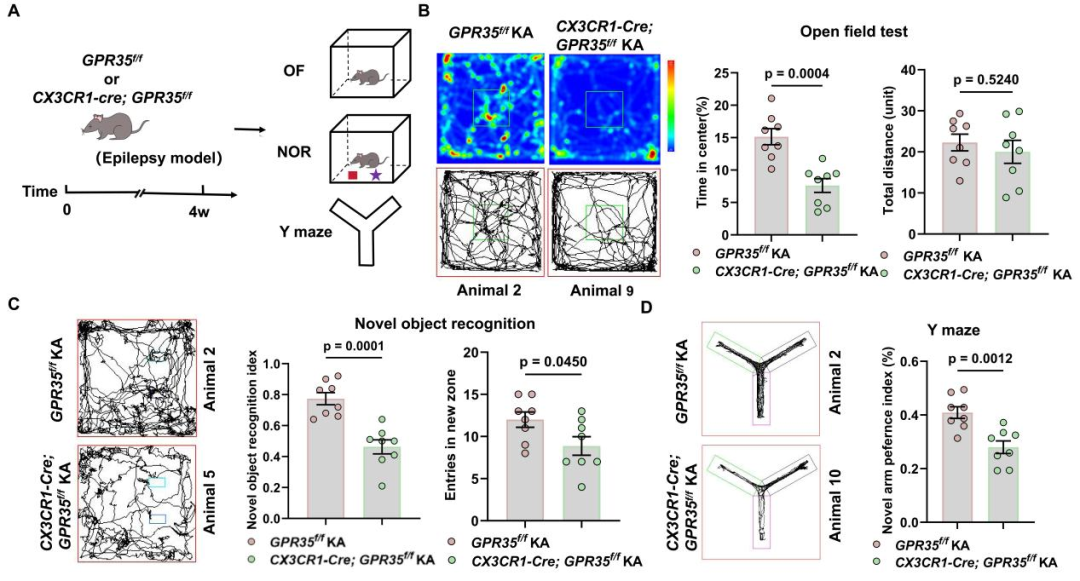
随后,研究人员评估小胶质细胞特异性GPR35缺失对癫痫小鼠认知功能的影响。结果显示,与GPR35f/f癫痫小鼠相比,CX3CR1-Cre;GPR35f/f癫痫小鼠运动距离无明显差异,但不愿探索中央区域,中央区域停留时间占比降低,提示焦虑样行为增强。与对照癫痫小鼠相比,CX3CR1-Cre;GPR35f/f癫痫小鼠在NOR与Y迷宫实验中均表现出认知偏好受损,提示空间工作记忆障碍。
进一步评估L-Kyn对CX3CR1-Cre;GPR35f/f癫痫小鼠认知功能的影响。与经PBS处理的GPR35f/f癫痫小鼠比较,OFT中经L-Kyn处理的GPR35f/f癫痫小鼠在中央区域停留时间占比显著升高;NOR与Y迷宫实验中,该组小鼠表现出明显偏好;MWM中,该组小鼠穿越原平台位置的次数更多,且在目标象限停留时间占比显著升高。但L-Kyn未能在CX3CR1-Cre;GPR35f/f癫痫小鼠中减轻焦虑样行为、改善空间工作记忆损伤。综合这些行为学数据可见,给予L-Kyn无法恢复CX3CR1-Cre;GPR35f/f癫痫小鼠的认知功能。总体而言,L-Kyn可通过激活GPR35抑制小鼠癫痫发作并改善癫痫相关认知损伤,而在小胶质细胞特异性GPR35敲除小鼠中,上述保护作用消失。
图11.L-Kyn可缓解KA诱导的GPR35f/f小鼠癫痫发作,并改善WT癫痫小鼠认知损伤
图12.小胶质细胞特异性GPR35缺失加剧KA诱导癫痫小鼠认知功能损伤
图13.L-Kyn不能改善KA诱导的CX3CR1-Cre;GPR35 f/f小鼠的认知功能障碍
结论
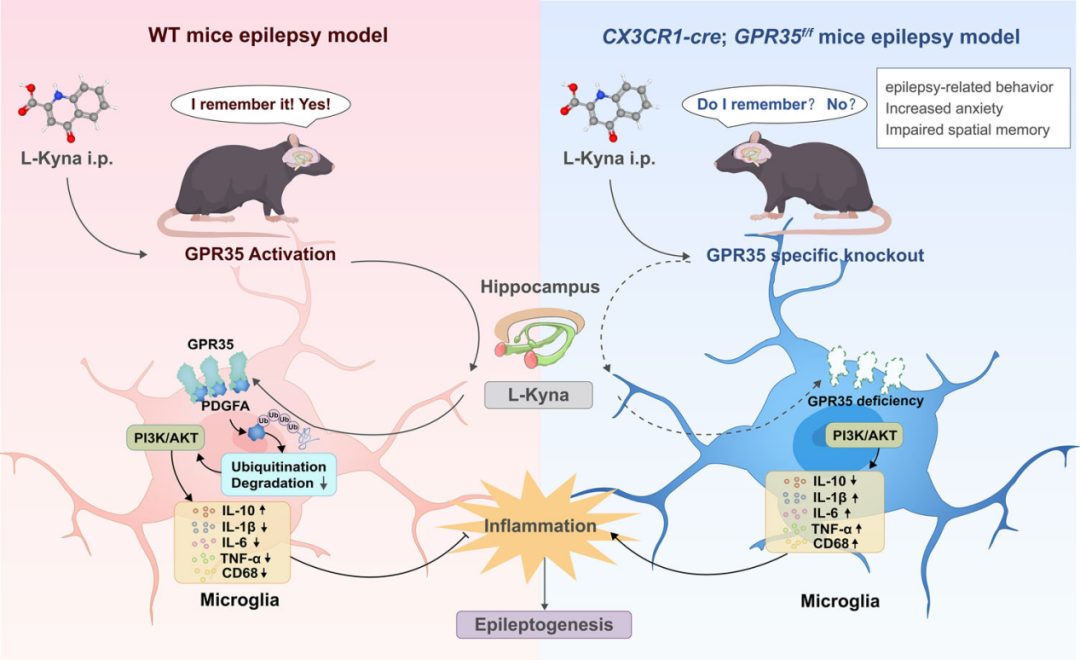
本研究结果表明,GPR35可抑制神经炎症、缓解癫痫发生过程。机制上,GPR35与PDGFA相互作用,并通过调控泛素—蛋白酶体途径稳定PDGFA,进而延缓癫痫进展。本研究首次阐明了GPR35参与神经炎症与癫痫发病的关键分子机制,为癫痫治疗提供新的干预思路和潜在治疗靶点。
示意图:小胶质细胞GPR35可通过调控泛素—蛋白酶体途径稳定PDGFA,抑制癫痫小鼠海马区神经炎症级联反应,进而缓解癫痫发生与进展。
本文使用的病毒产品,列表如下:
扫码添加客服

更多产品及详情欢迎咨询!

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK