2026-02-24 阅读量:274
近日,南京中医药大学郑仕中教授团队,在International Journal of Biological Sciences(IF=10.0)发表研究成果“Curcumol Induces Necroptosis of Hepatic Stellate Cells by Targeting KAT8 to Suppress HK2 Lactylation and Promote HUWE1-Dependent Ubiquitination”,创新性地揭示了肝纤维化中肝星状细胞(HSC)存活的新代谢调控机制:糖酵解的产物乳酸通过激活乳酸化转移酶KAT8,催化糖酵解关键酶HK2发生乳酸化修饰,从而稳定HK2蛋白并维持细胞活化。
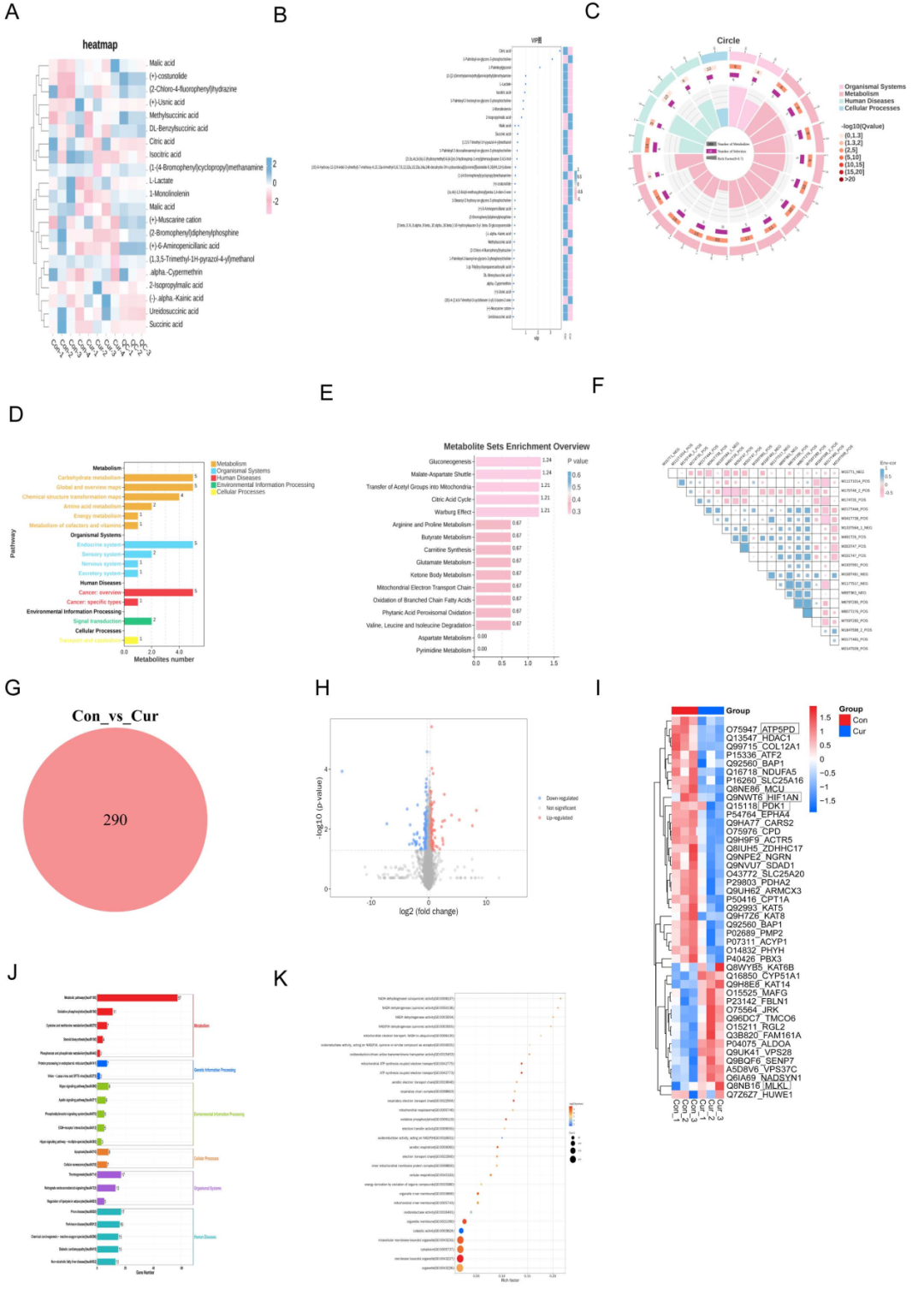
研究人员通过对莪术醇处理的人肝星状细胞系(LX-2)进行蛋白质组学与非靶向代谢组学分析,发现莪术醇破坏细胞能量代谢并诱导LX-2细胞发生坏死性凋亡。代谢组数据显示,莪术醇显著影响糖酵解和三羧酸循环中的多个关键代谢物。对乳酸、柠檬酸和苹果酸等差异代谢物的分析,结合VIP分析、表达热图以及KEGG通路富集结果,共同凸显了糖酵解(Warburg效应)与线粒体能量代谢重编程在肝星状细胞中的关键作用。此外,相关性分析表明代谢物谱的变化与细胞能量失衡之间存在直接关联。
同时,蛋白质组学分析提供了补充证据,维恩图和火山图定量描述了蛋白质表达谱的变化。这些结果揭示了参与葡萄糖代谢、线粒体能量生成和坏死性凋亡的蛋白质发生显著改变。此外,通路富集分析表明与线粒体氧化磷酸化和脂肪酸代谢相关的蛋白质显著富集。基因本体(GO)分析显示差异表达的基因和蛋白质主要富集于线粒体能量代谢通路。如图1K所示,这些过程包括电子传递链和ATP合成等。基于以上发现,研究人员提出莪术醇可能破坏糖酵解与线粒体代谢之间的耦合,导致细胞能量崩溃。
图1.多组学分析揭示了莪术醇诱导肝星状细胞代谢功能障碍与坏死性凋亡的过程。
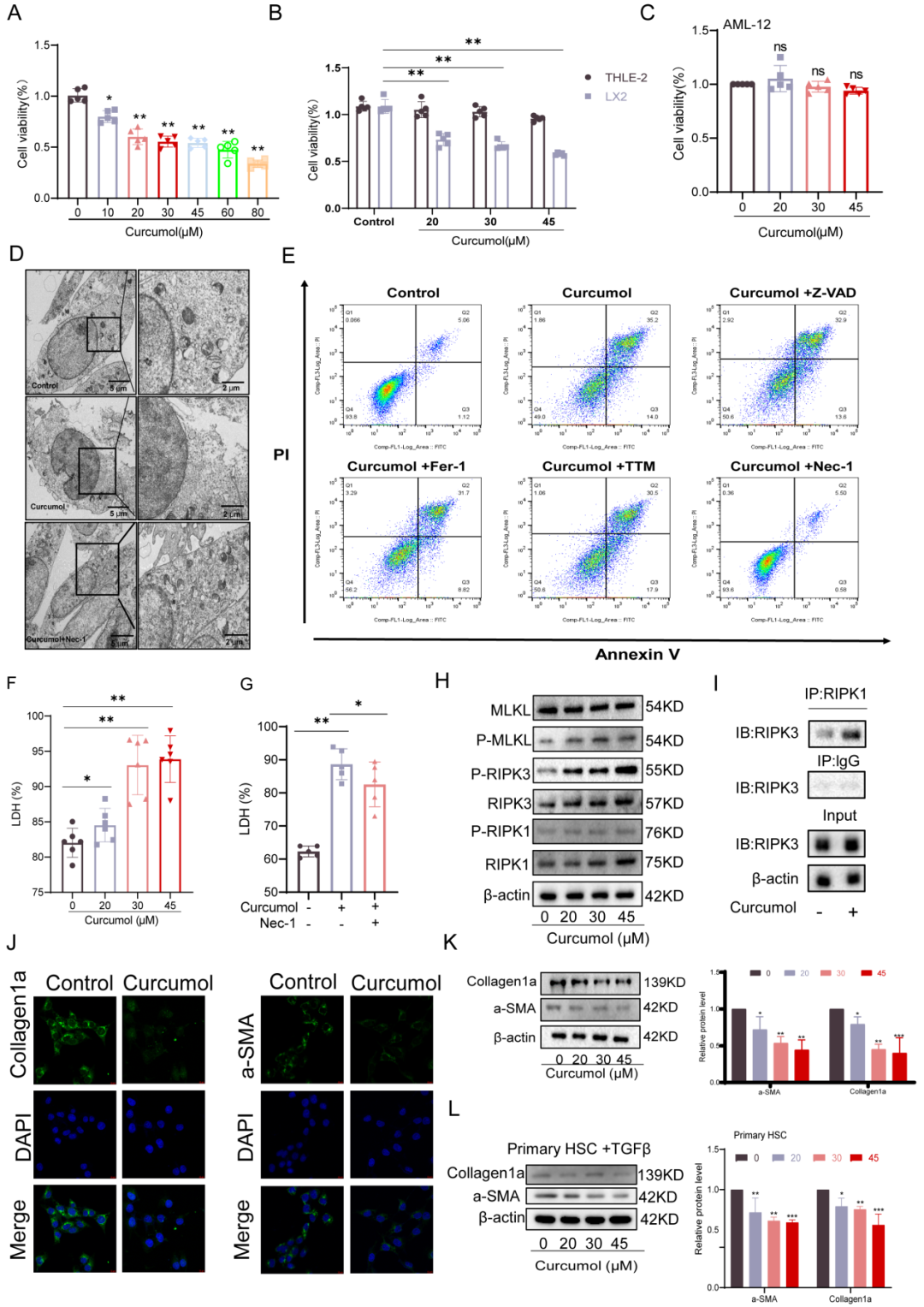
接着为确定莪术醇处理LX-2细胞的最佳浓度,研究人员首先评估了不同浓度(0-80 μM)处理24小时后对细胞活力的影响。结果表明,适用于后续实验的有效浓度范围为0-45 μM。除细胞活力下降外,莪术醇处理的LX-2细胞中乳酸脱氢酶(LDH)水平显著升高,提示细胞膜完整性丧失并发生细胞死亡。
为探究莪术醇诱导LX-2细胞死亡的机制,研究人员基于多组学结果及前期研究检测了坏死性凋亡通路。坏死性凋亡特异性抑制剂Necrostatin-1(Nec-1)处理显著逆转了透射电镜下观察到的莪术醇诱导的细胞死亡相关形态改变。流式分析进一步表明,Nec-1显著降低了莪术醇诱导的LX-2细胞晚期凋亡与坏死细胞比例。此外,Nec-1明显抑制了莪术醇引起的LDH释放增加。相比之下,其他细胞死亡抑制剂,包括凋亡抑制剂Z-VAD-FMK(Z-VAD)、铁死亡抑制剂Ferrostatin-1(Fer-1)与Liproxstatin-1(Lipo-1)以及铜死亡抑制剂Tetrathiomolybdate(TTM),均未对莪术醇引起的LDH释放产生显著影响。
在机制层面,Western blot分析显示,随着莪术醇浓度升高,LX-2细胞中受体相互作用丝氨酸/苏氨酸蛋白激酶RIPK1、RIPK3、混合谱系激酶结构域样蛋白(MLKL)及其磷酸化形式(p-RIPK1、p-RIPK3、p-MLKL)的表达呈剂量依赖性上升,这些结果证实莪术醇激活了经典的RIPK1/RIPK3介导的坏死性凋亡信号通路。此外,免疫共沉淀实验表明,莪术醇处理增强了RIPK1与RIPK3之间的相互作用,这是启动坏死性凋亡的关键事件。
研究人员进一步评估了莪术醇对HSC活化标志物的影响。免疫荧光染色与Western blot分析显示,莪术醇处理显著降低了LX-2细胞中Ⅰ型胶原蛋白α链(Collagen1a)与α-平滑肌肌动蛋白(α-SMA)的表达,在原代小鼠HSC中也得到类似结果。综上,莪术醇通过激活RIPK1/RIPK3信号通路、促进坏死小体复合物形成,诱导LX-2细胞发生坏死性凋亡,并选择性抑制HSC的活化与增殖。
图2.莪术醇诱导HSC发生坏死性凋亡。
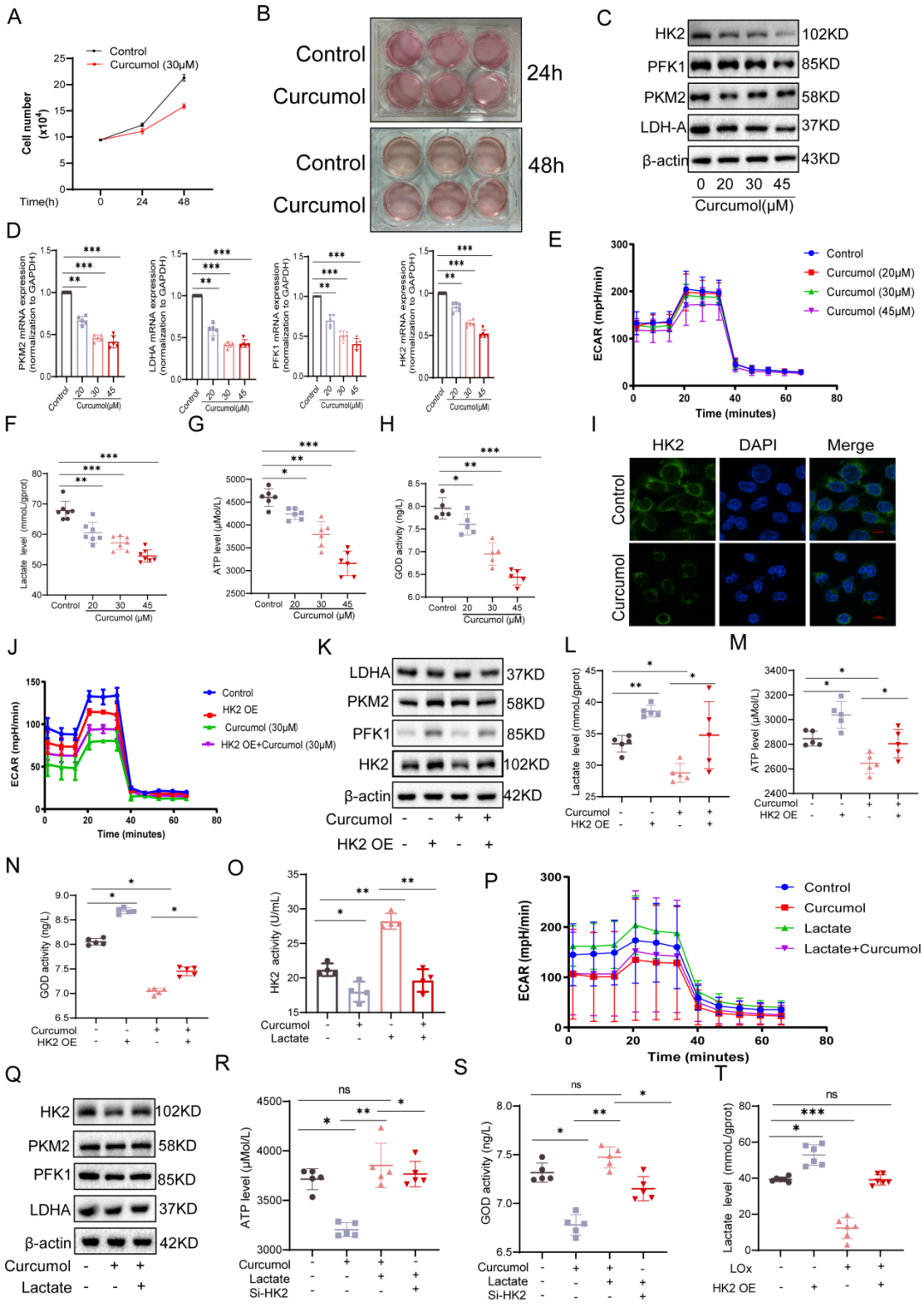
在研究莪术醇对肝星状细胞(HSC)作用机制的过程中,研究人员观察到对照组培养基比莪术醇处理组更快变黄。结合代谢组学结果,这一现象提示莪术醇可能干扰了细胞糖酵解过程。蛋白质印迹分析证实,莪术醇处理可浓度依赖性(0-45 μM)降低HSC中关键糖酵解酶(包括HK2、PKM2、PFK1和LDHA)的蛋白表达。
为进一步评估LX-2细胞的糖酵解活性,研究人员使用Seahorse XF分析仪进行了代谢图谱分析。结果显示,莪术醇处理可剂量依赖性地降低细胞外酸化率(ECAR),同时减少乳酸生成、降低ATP水平并减少葡萄糖消耗。以上结果表明,莪术醇通过下调关键糖酵解酶的表达与活性,显著降低了LX-2细胞的糖酵解能力。其中,HK2似乎是受影响最显著的目标。前期研究已证实,HSC特异性或系统性HK2缺失均能抑制HSC活化并减轻体内肝纤维化。因此,莪术醇很可能主要通过调控HK2来调节HSC的糖酵解过程。免疫荧光分析显示,莪术醇处理降低了HSC中HK2的表达,进一步验证了莪术醇通过抑制HK2来调控HSC糖酵解。
利用HK2过表达的LX-2细胞模型,研究人员发现莪术醇处理后,HK2过表达细胞的ECAR仍与对照组相当。此外,HK2过表达显著逆转了莪术醇对多个糖酵解指标的抑制作用,包括糖酵解酶表达、乳酸生成、ATP合成以及葡萄糖消耗。这些结果表明,莪术醇主要通过抑制HK2蛋白功能来降低HSC的糖酵解通量。HK2过表达可逆转该效应,进一步证实莪术醇主要通过功能性抑制HK2来调控HSC的代谢重编程。
在明确莪术醇通过靶向HK2抑制HSC糖酵解后,研究人员进一步探究了其上游调控机制。活化的HSC表现为糖酵解活性增强、乳酸累积增加以及HK2表达异常升高。越来越多的研究表明,乳酸不仅是代谢副产物,还可作为信号分子调控多种蛋白活性。然而,乳酸是否直接调控HK2仍不明确。
本研究中,研究人员发现外源性乳酸的添加能有效缓解30 μM莪术醇对HK2活性的抑制作用,并恢复关键糖酵解酶的表达。此外,乳酸补充还逆转了莪术醇引起的ECAR下降、ATP生成减少以及葡萄糖消耗降低。为进一步确认乳酸的作用,研究人员使用乳酸氧化酶(LOx)降解细胞内乳酸,该处理显著降低HSC内乳酸水平,并有效抑制了HK2过表达诱导的乳酸累积。综上所述,这些结果表明乳酸对维持HSC中HK2功能及糖酵解活性至关重要,提示乳酸是HSC活化相关代谢重编程中的关键代谢信号媒介。
图3.莪术醇通过抑制乳酸-HK2正反馈环路,调控HK2功能,从而抑制肝星状细胞的糖酵解过程。
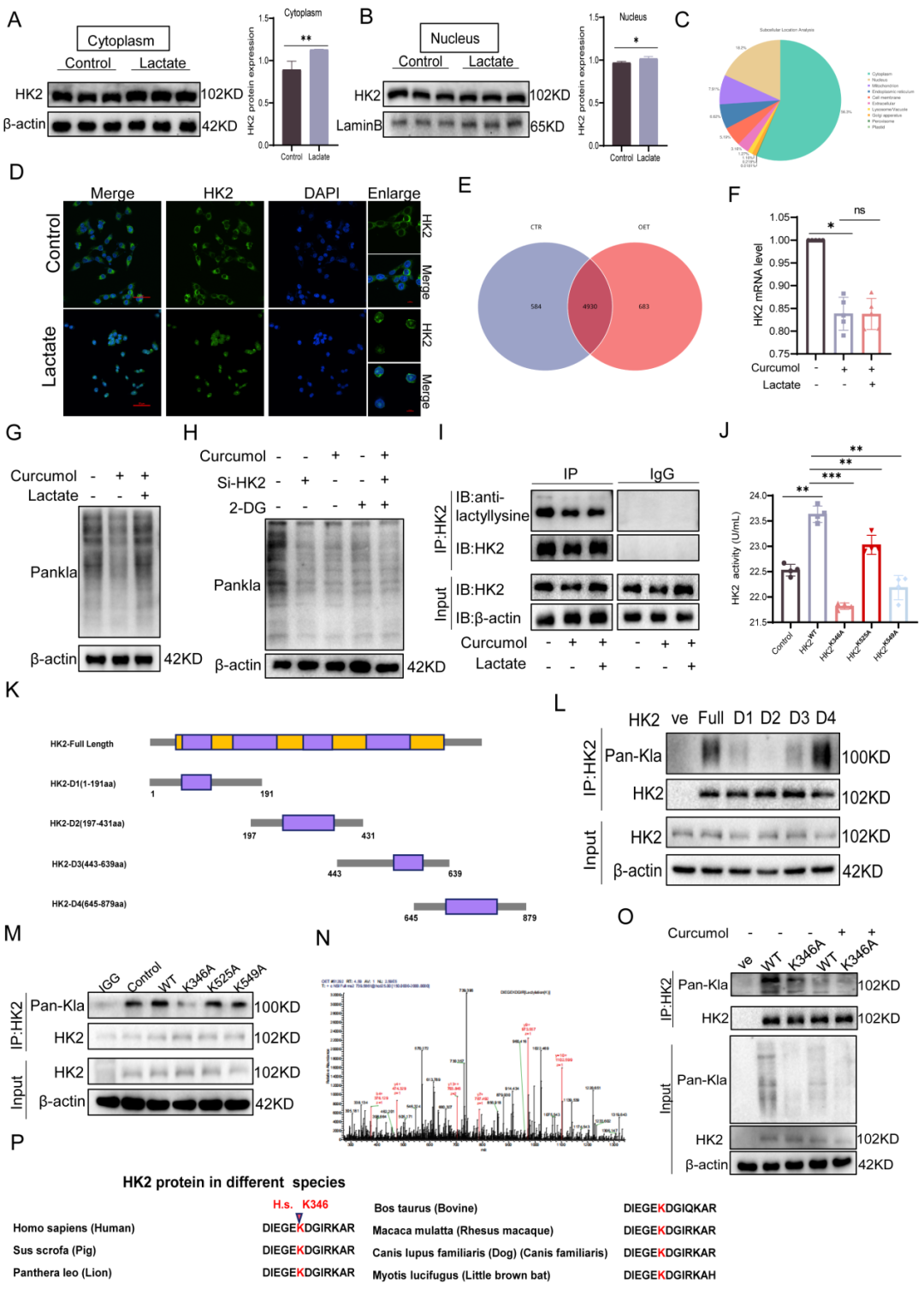
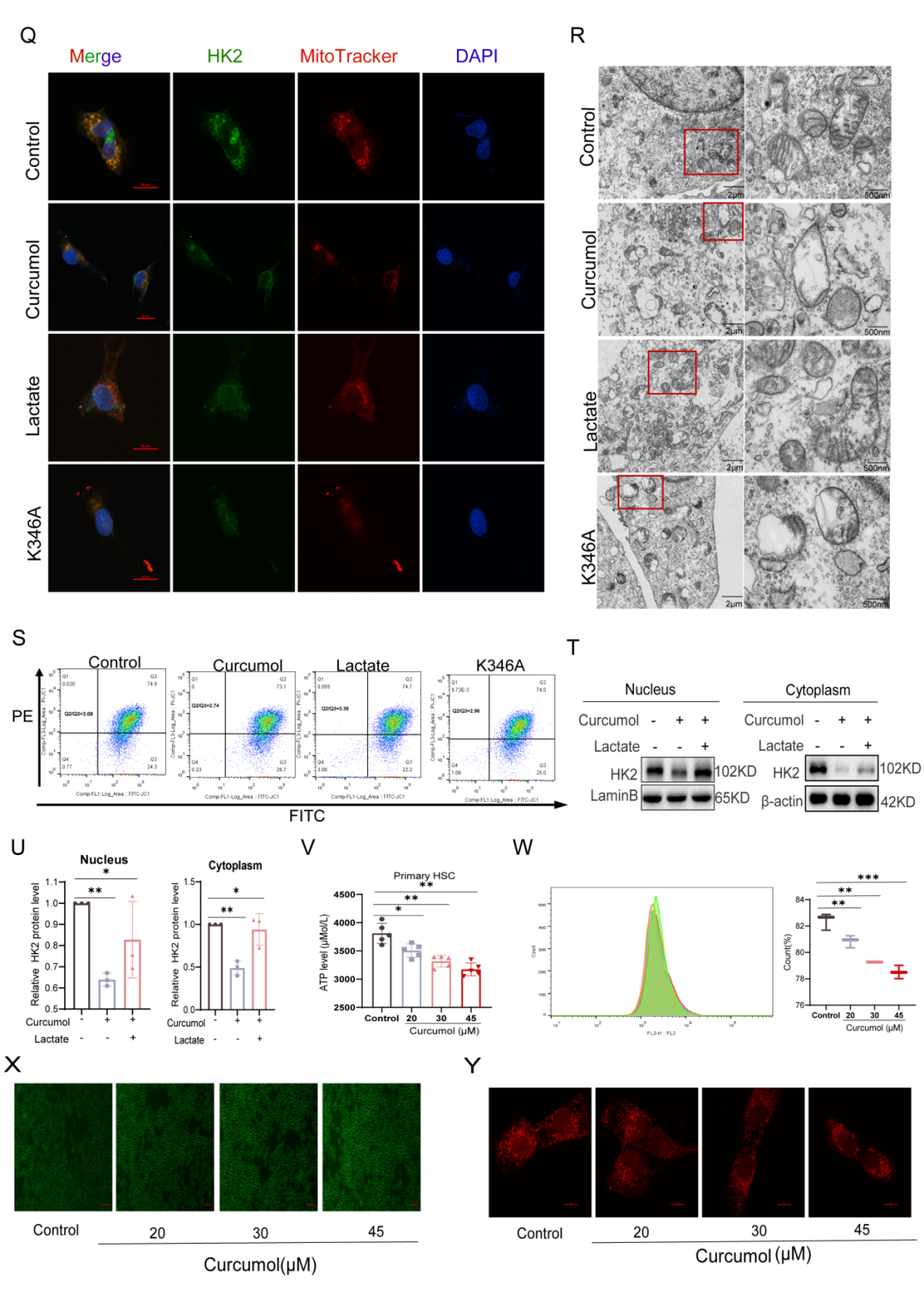
为进一步探究莪术醇是否通过乳酸介导的翻译后修饰调控HK2,研究人员开展了一系列实验。首先,外源性乳酸钠处理显著促进了HK2的核转位,并提升了其胞浆蛋白水平,但HK2 mRNA表达未见明显变化。这些结果表明乳酸通过翻译后机制而非转录调控来调节HK2。
此外,免疫沉淀-质谱(IP-MS)分析显示,乳酸化修饰蛋白主要定位于细胞质,同时在细胞核和线粒体中也有富集。蛋白质印迹分析发现,莪术醇处理降低了细胞内整体蛋白质乳酸化水平,而外源性乳酸补充可部分恢复该修饰。同时,联合使用莪术醇、糖酵解抑制剂2-DG及HK2 siRNA进一步抑制了蛋白质乳酸化。Co-IP实验直接证实莪术醇显著抑制HK2的乳酸化修饰。
为确定具体的修饰位点,研究人员构建了HK2截短突变体,将乳酸化修饰定位至第二(D2)结构域。随后的定点突变显示K346为关键的乳酸化位点,而K525A与K549A突变影响甚微。K346突变不仅大幅降低HK2乳酸化修饰,还损害了其酶活性。序列比对表明该位点在物种间高度保守。重要的是,莪术醇抑制野生型HK2(HK2-WT)的乳酸化修饰,但对K346A突变体影响很小,表明莪术醇特异性靶向HK2 K346位点的乳酸化修饰。
免疫荧光分析进一步证实,莪术醇减少了HK2在线粒体和细胞核中的定位,亚细胞分级分离实验也支持这一结论。乳酸补充完全恢复了HK2的核转位,而K346A突变体再现了莪术醇的效应,提示K346位点的乳酸化是控制HK2亚细胞定位的分子开关。
在细胞器水平上,透射电镜观察显示莪术醇处理后线粒体出现广泛损伤,包括嵴结构破坏和基质肿胀。尽管乳酸补充或HK2 K346突变未能逆转这些结构异常,但它们部分缓解了莪术醇诱导的线粒体膜电位丧失。虽然乳酸恢复了HK2定位,但并未逆转结构损伤,表明乳酸化修饰除调控亚细胞定位外,还影响线粒体完整性。
进一步实验表明,莪术醇显著增加了线粒体活性氧水平,改变了HK2在细胞质与细胞核之间的分布,降低了ATP生成,减少了线粒体数量,并破坏了线粒体膜电位。
综上,莪术醇选择性抑制HK2 K346位点的乳酸化修饰,阻止其核转位与线粒体定位,这一破坏导致线粒体功能障碍和氧化应激,最终瓦解活化肝星状细胞的稳态。
图4.莪术醇通过抑制HK2蛋白第346位赖氨酸的乳酸化修饰,阻断其核转位并诱导线粒体功能障碍。
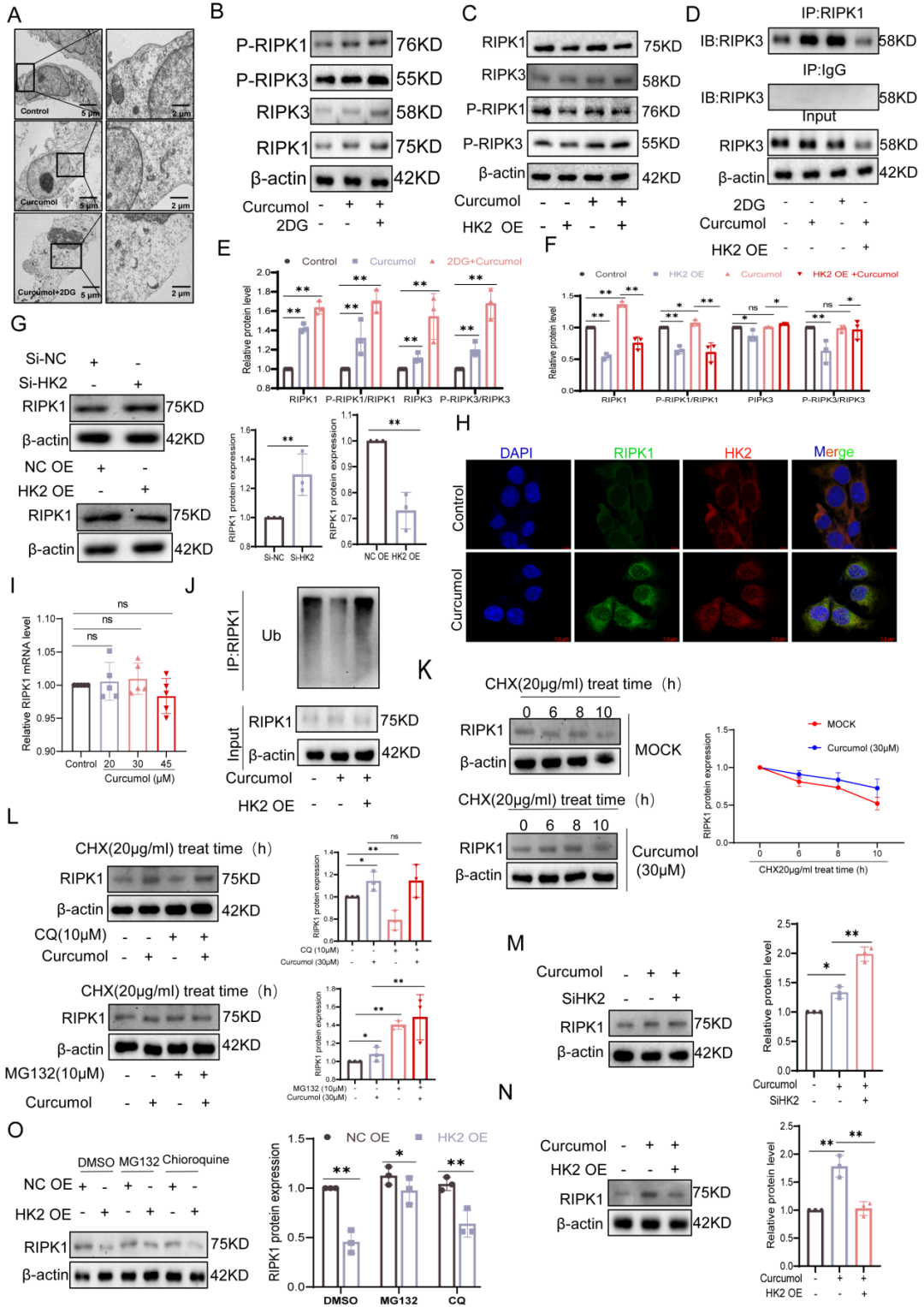
为评估莪术醇对LX-2细胞坏死性凋亡的影响及HK2在其中的作用,透射电镜观察显示,24小时莪术醇处理可诱发典型的坏死特征,包括质膜破裂和细胞器肿胀。该效应被糖酵解抑制剂2-DG显著增强,提示代谢应激与坏死性凋亡信号间存在协同作用。
在分子水平上,莪术醇特异性激活了RIPK1/RIPK3磷酸化级联反应,而HK2过表达则完全阻断了这一激活过程,证实HK2是坏死性凋亡的关键调控节点。在过表达HK2的LX-2细胞中,免疫共沉淀实验显示HK2促进了RIPK1与RIPK3之间的相互作用。蛋白质印迹分析进一步表明,HK2过表达显著降低了RIPK1蛋白水平,而使用siRNA敲低HK2则部分恢复了RIPK1的表达。
为排除转录调控的影响,研究人员检测了HK2过表达或敲低后RIPK1 mRNA水平的变化。结果显示,改变HK2表达并未显著影响RIPK1 mRNA水平,进一步证明HK2介导的RIPK1表达变化主要发生在翻译后水平。与此一致,免疫荧光分析显示莪术醇处理后细胞内HK2与RIPK1的共定位增加。
考虑到泛素化在控制蛋白稳定性中的关键作用,研究人员推测莪术醇可能通过HK2影响RIPK1的泛素化,进而调控其稳定性。免疫沉淀实验支持这一假设:在过表达HK2的莪术醇处理LX-2细胞中,RIPK1泛素化水平显著降低。为深入探究HK2在此过程中的具体作用,研究人员构建了HK2 K346位点突变体并进行泛素化分析。结果显示,K346位点突变有效抑制了RIPK1泛素化,表明K346是HK2介导RIPK1泛素化调控的关键位点。
RT-qPCR分析排除了转录调控的可能性,因为莪术醇处理并未显著改变RIPK1 mRNA水平。蛋白稳定性实验显示,莪术醇明显延长了RIPK1的半衰期。此外,放线菌酮(CHX)追踪实验和蛋白酶体抑制剂MG132处理证实,RIPK1稳定性受泛素-蛋白酶体途径调控。与此一致,自噬抑制剂氯喹(CQ)与莪术醇共处理HSC的结果显示,抑制自噬并未改变莪术醇诱导的RIPK1蛋白水平上调。同时,对自噬流标志物LC3-II/I和p62的评估表明,莪术醇对HSC的基础自噬无显著影响。这些发现进一步支持了HK2通过泛素-蛋白酶体途径调控RIPK1的结论。
值得注意的是,该调控作用依赖于HK2:敲低HK2显著降低了RIPK1蛋白水平,而过表达HK2则抵消了莪术醇对RIPK1降解的促进作用。综上,这些结果表明莪术醇通过HK2抑制RIPK1的泛素化,从而稳定RIPK1蛋白,激活坏死性凋亡信号通路,诱导LX-2细胞发生坏死性凋亡。
图5.莪术醇通过抑制HK2介导的RIPK1泛素化,诱导肝星状细胞发生坏死性凋亡。
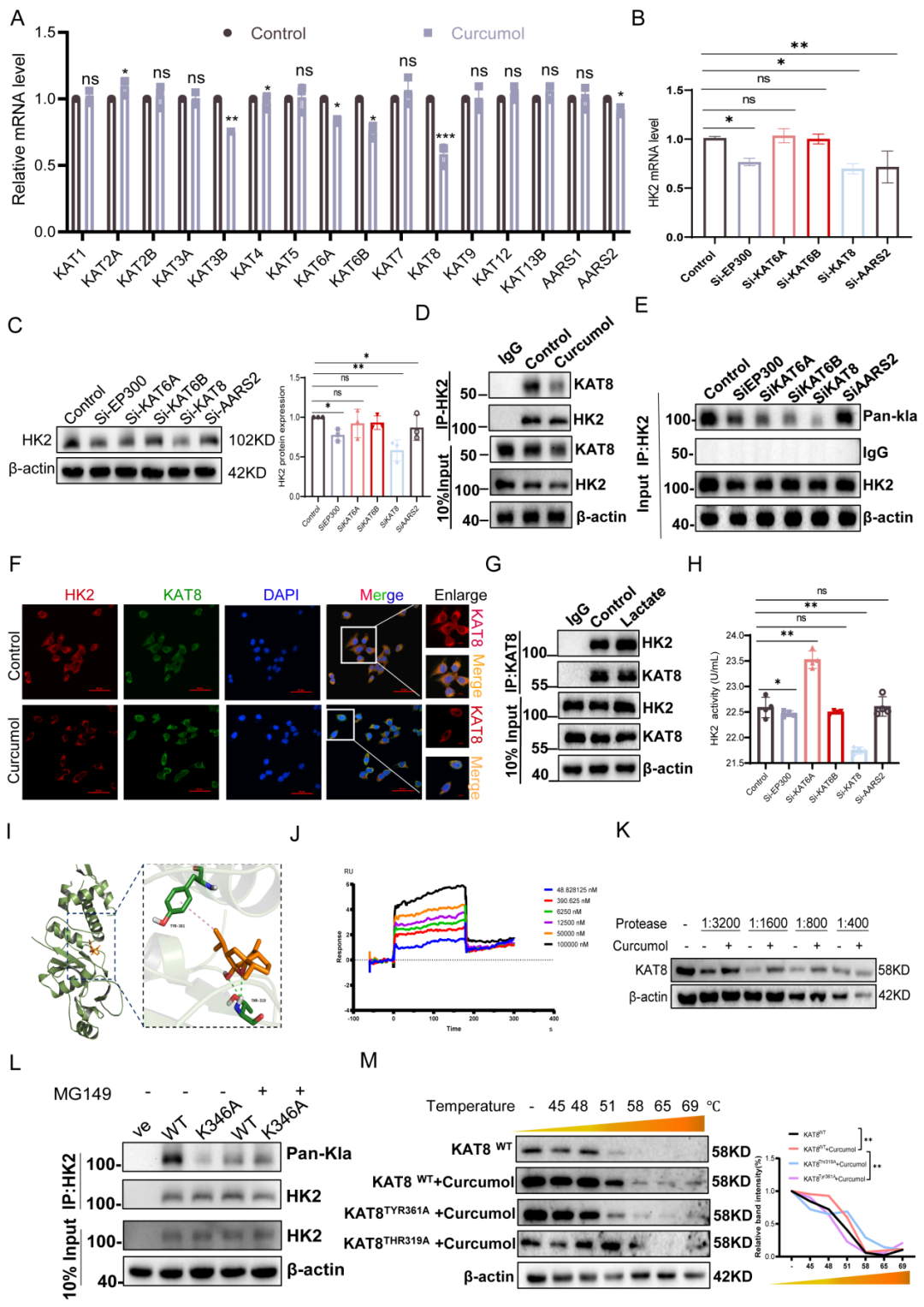
为探究莪术醇调控HK2乳酸化修饰的分子机制,研究人员采用高通量RT-qPCR检测了其对LX-2细胞中乳酸化相关酶表达的影响。处理24小时后,KAT3B(EP300)、KAT6A、KAT6B、KAT8和AARS2的mRNA水平均显著降低。为确定关键调控因子,研究人员进行了siRNA介导的敲低实验。结果显示,沉默KAT8能显著降低HK2的mRNA和蛋白表达水平,而敲低其他候选基因影响甚微,表明KAT8在调控HK2表达中具有特异且关键的作用。
在翻译后水平上,KAT8敲低显著降低了HK2乳酸化修饰,而其他siRNA组的敲低效果不明显。同样,KAT8特异性抑制剂MG149处理也抑制了HK2乳酸化,且该效应在HK2 K346突变体中完全消失,直接证实KAT8是催化该位点乳酸化修饰的关键酶。免疫共沉淀实验进一步表明,莪术醇处理减弱了KAT8与HK2之间的相互作用。免疫荧光分析进一步确认了两者在细胞内的共定位,且该共定位在莪术醇处理后被打乱。
外源性乳酸的添加增强了KAT8与HK2的互作,提示乳酸可能作为信号分子促进两者的结合并推动HK2乳酸化修饰。酶活测定显示,KAT8敲低显著降低HK2活性,而其他siRNA组敲低影响较小。
为探究莪术醇是否直接靶向KAT8,研究人员首先进行了分子对接分析,结果显示莪术醇与KAT8存在潜在相互作用。随后采用药物亲和反应靶点稳定性(DARTS)实验,该实验基于药物结合后靶蛋白对蛋白酶解的抗性增强的原理。结果显示莪术醇处理显著提高了KAT8对抗蛋白酶解的稳定性,为两者相互作用提供了直接生化证据。进一步通过表面等离子共振分析定量验证了该结合,表明莪术醇与固定化KAT8之间存在高亲和力相互作用。
在此基础上,研究人员通过细胞热转移实验对精确结合位点进行定位,发现TYR361A和THR319A突变体与莪术醇的结合亲和力较野生型KAT8显著降低。最后,莪术醇处理增强了KAT8与HK2之间的相互作用。
综上所述,这些结果证明KAT8直接催化HK2 K346位点的乳酸化修饰,而莪术醇通过直接结合KAT8抑制该修饰,从而破坏HK2介导的代谢重编程。
图6.KAT8介导HK2蛋白K346位点的乳酸化修饰。
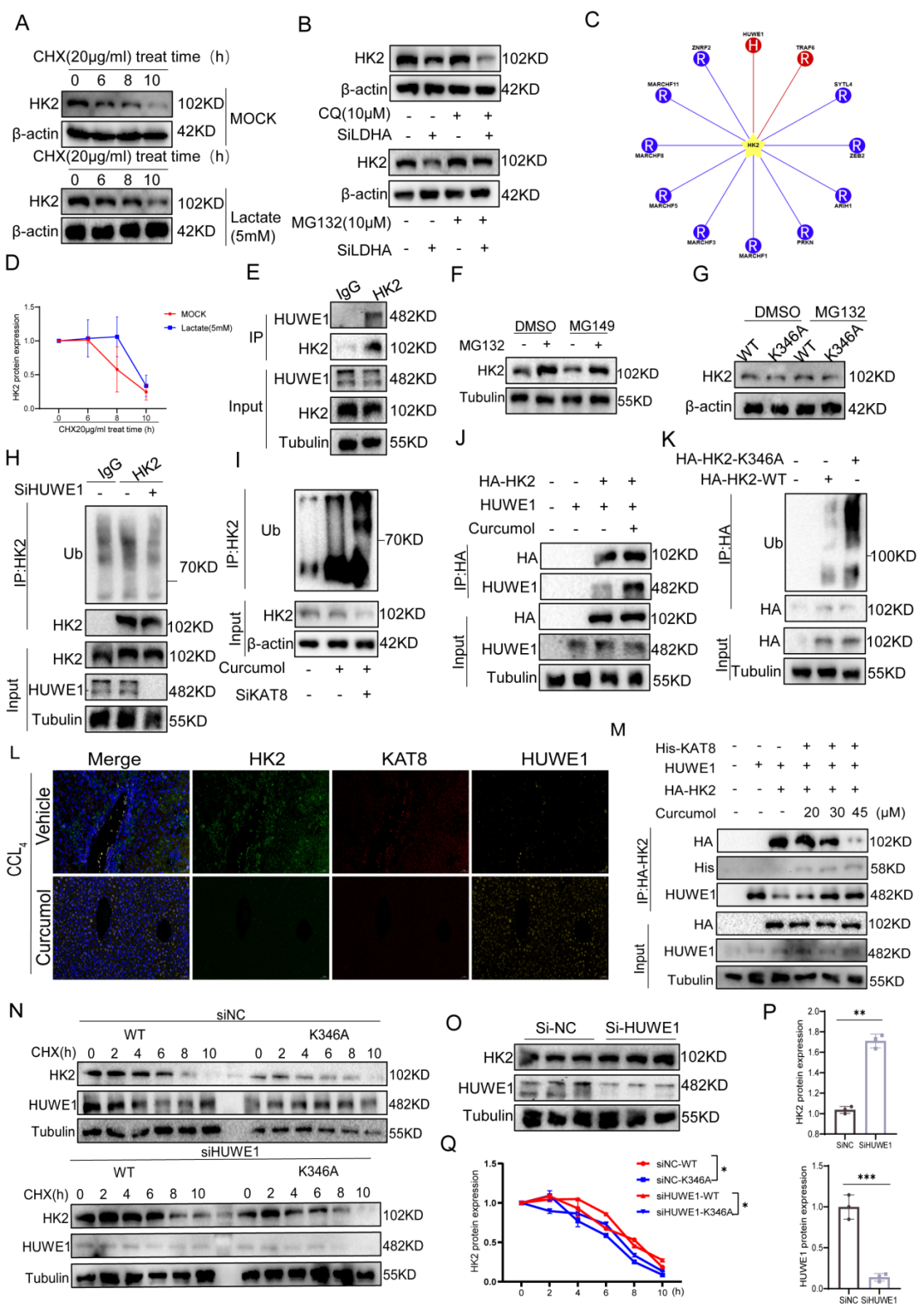
为探究乳酸调控HK2蛋白稳定性的作用,研究人员首先进行了蛋白半衰期实验。使用5 mM乳酸处理LX-2细胞24小时显著延缓了HK2的降解。在蛋白质合成受抑制的条件下,密度分析证实HK2的半衰期明显延长,表明乳酸通过抑制蛋白降解来稳定HK2。
进一步实验发现,敲低LDHA显著降低了HK2蛋白水平,而蛋白酶体抑制剂MG132能完全挽救这一下降,自噬抑制剂CQ则无效,表明HK2的降解依赖于蛋白酶体途径,且需要LDHA介导的乳酸生成。通过E3泛素连接酶筛选,研究人员确定HECT家族E3(HUWE1)是负责HK2泛素化的主要连接酶,免疫共沉淀实验证实了HK2与HUWE1的直接相互作用。
进一步分析显示,使用MG149抑制乳酸化酶KAT8显著提高了HK2蛋白水平,而蛋白酶体抑制剂MG132处理则阻断了由KAT8抑制引发的HK2降解(图7F)。此外,莪术醇处理和KAT8敲低均增加了HK2的泛素化,并表现出协同效应(图7I),提示莪术醇可能通过抑制KAT8依赖的乳酸化修饰来促进HUWE1介导的泛素化。
在蛋白酶体抑制条件下,野生型HK2明显积累,而K346A突变体未出现显著积累。泛素化分析显示,K346A突变体的泛素化水平显著高于野生型HK2。值得注意的是,敲低HUWE1显著提高了野生型HK2的稳定性,但对K346A突变体无影响。在HUWE1缺陷细胞中,K346A突变体仍持续快速降解。这些结果表明,在调控HK2稳定性方面,KAT8介导的K346乳酸化修饰与HUWE1驱动的泛素化之间存在竞争性关联。
在HEK293T细胞中的过表达实验显示,过表达KAT8会破坏HUWE1与HK2之间的相互作用,而莪术醇则促进两者的结合。免疫荧光分析显示,在肝组织中HK2、KAT8和HUWE1存在共定位,且在纤维化小鼠肝脏中HK2与KAT8的共定位显著增强,表明该调控通路在病理状态下被激活。
综上所述,这些发现表明乳酸通过激活KAT8催化HK2 K346位点的乳酸化修饰,从而削弱HUWE1介导的泛素化并阻止HK2的蛋白酶体降解。莪术醇则通过抑制KAT8介导的乳酸化修饰,恢复HUWE1活性,并促进泛素依赖的HK2降解,进而逆转这一效应。
图7.莪术醇通过抑制KAT8介导的K346位点乳酸化修饰,促进HUWE1依赖的泛素化降解HK2蛋白。
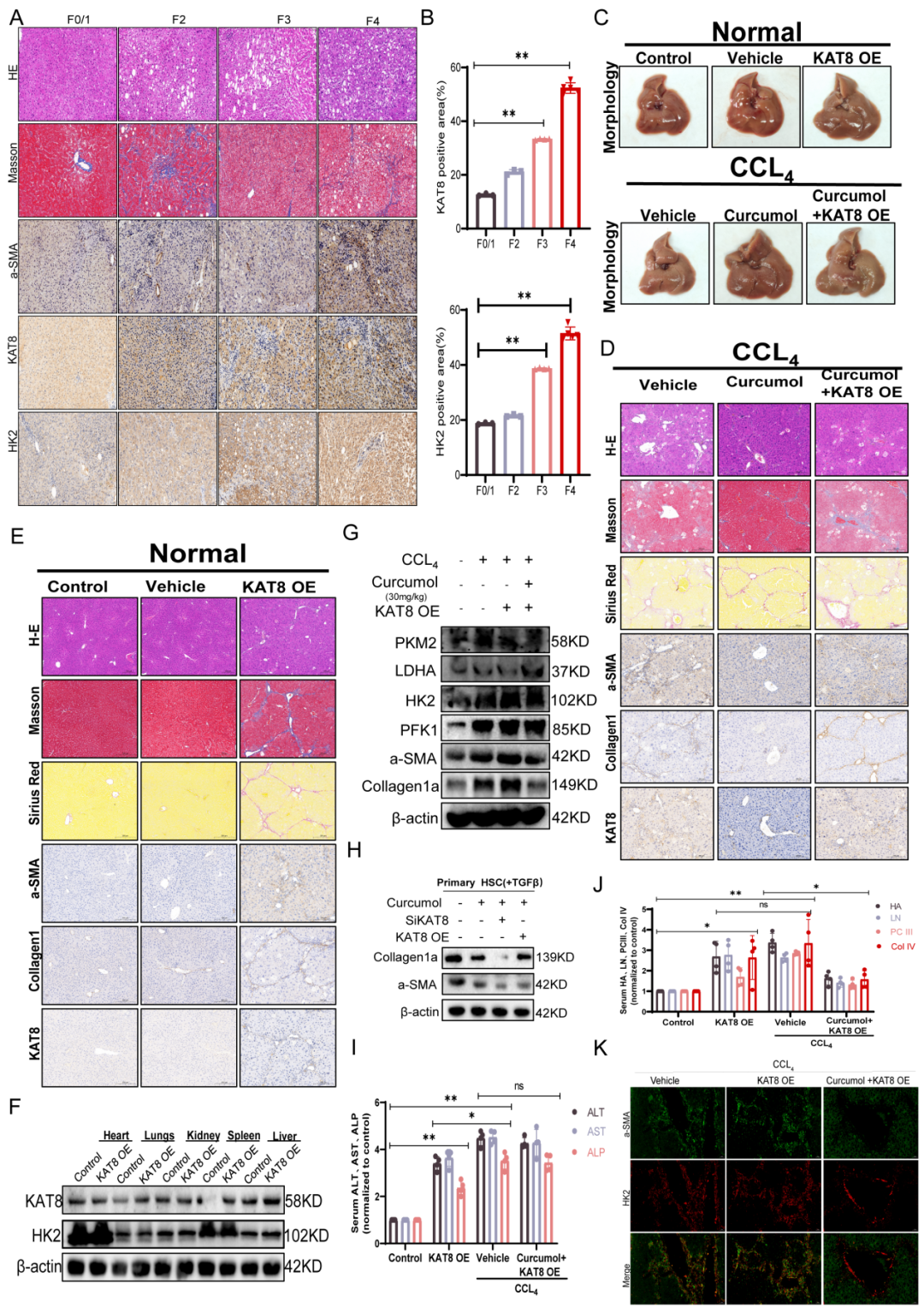
为深入探究KAT8与HK2在肝纤维化病理进展中的作用,研究人员检测了二者在临床人肝纤维化组织样本中的表达情况。通过H&E染色、Masson染色及α-SMA(活化的肝星状细胞标志物)免疫组化的组织学分析,结果显示患者肝组织呈现典型的纤维化特征。随后基于Metavir评分系统进行的半定量分析表明,纤维化肝组织中KAT8与HK2的表达呈显著正相关。
机制研究表明,KAT8过表达显著加剧了CCl4诱导的肝纤维化,表现为:肝脏/体重比增加;通过H&E、Masson和天狼星红(Sirius red)染色评估的胶原沉积增多;α-SMA和I型胶原表达上调,以及血清纤维化和肝损伤标志物水平升高。相反,莪术醇处理减轻了这些病理改变,但KAT8过表达完全抵消了其保护作用,这突显了KAT8在纤维化进程中的关键作用。
进一步分析显示,KAT8过表达提高了肝脏HK2蛋白水平,上调了糖酵解酶及纤维化相关蛋白的表达,这些结果在原代HSC中得到证实。与此相伴的是血清乳酸水平显著升高。免疫荧光分析进一步表明,纤维化肝脏中α-SMA与HK2的共定位增强,莪术醇处理可减少这种共定位,而KAT8过表达则使其完全恢复。
总之,体内外实验结果共同表明,莪术醇通过靶向KAT8抑制HSC糖酵解,减轻HK2介导的坏死性凋亡信号抑制,并激活RIPK1/RIPK3通路诱导坏死性凋亡,从而产生强大的抗纤维化作用。
图8.KAT8通过HK2介导的RIPK1依赖性坏死性凋亡调控,在体内促进肝纤维化。
总结
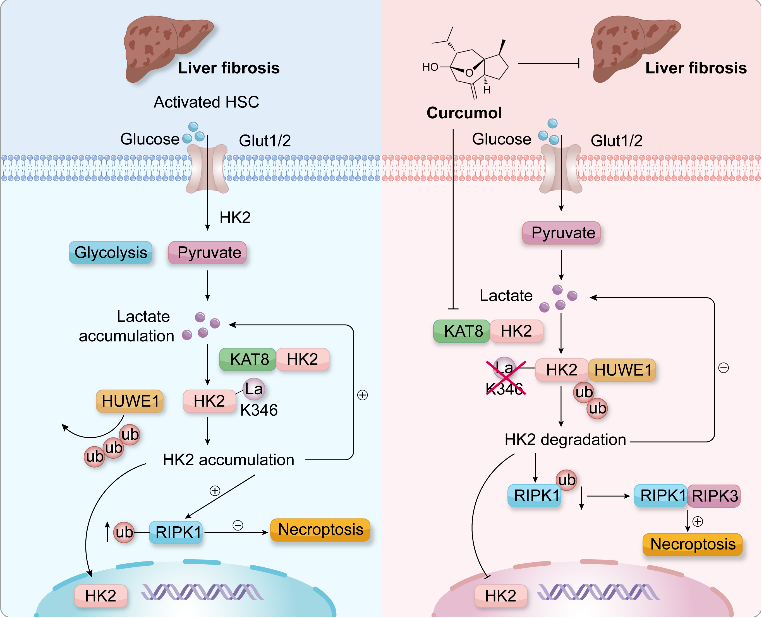
该研究从代谢重编程出发,提出HK2乳酸化修饰是调控肝星状细胞存活的代谢关键节点,并确认莪术醇作为一种靶向KAT8-HK2通路的潜在抗纤维化化合物,成功将代谢抑制与坏死性凋亡细胞死亡机制相联系。
图9.全文机制总结
南京中医药大学硕士研究生郭晓涵为第一作者;南京中医药大学郑仕中教授为通讯作者。该研究得到国家自然科学基金等资助。
通讯介绍:
郑仕中,南京中医药大学教授、博士生导师。现任南京中医药大学药学院临床药学教研室主任,兼任江苏省中西医结合学会临床药学专业委员会副主任委员等学术职务。作为国家高层次人才计划引进学者,其研究方向聚焦中医药防治慢性肝病及肿瘤的作用机制,主持国家自然科学基金面上项目5项及其他课题20余项。在科研领域发表SCI论文180余篇,研究成果发表于《Hepatology》、《Autophagy》等国际顶刊,授权发明专利10项,主编国家级规划教材4部。
参考文献
[1]Guo X, Lin Y, Jiang Y, Wang M, Zheng S. Curcumol Induces Necroptosis of Hepatic Stellate Cells by Targeting KAT8 to Suppress HK2 Lactylation and Promote HUWE1-Dependent Ubiquitination. Int J Biol Sci. 2026 Jan 14;22(3):1648-1673. doi: 10.7150/ijbs.125009.
本文使用的病毒产品均来自枢密科技,列表如下:

扫码添加客服
更多产品及详情欢迎咨询!


市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK