2025-08-18 阅读量:707
高盐摄入是慢性非传染性疾病(尤其是心血管疾病)的主要饮食危险因素,在全球范围内,超过一半的饮食相关死亡都与高盐摄入有关。尽管世界卫生组织建议每日食盐摄入量应低于5克,但全球大多数人群的日均食盐消耗量仍超过10克。此外,过去20年间,尽管采取了多项减盐策略,人均每日盐摄入量却始终未发生改变。高盐摄入会导致离子稳态失衡、交感神经与肾素-血管紧张素-醛固酮系统(RAAS)激活、胰岛素抵抗等一系列病理生理变化,进而对心脏、大脑、肾脏、血管等多个器官造成损伤。因此,明确高盐摄入如何引发这些健康问题并开发有效的治疗方案,已成为当务之急。
组织内的过量盐分通过渗透压依赖的方式,促使促炎性免疫细胞富集。同时,高盐摄入还会直接导致肠道菌群失衡。肝脏作为营养物质进入血液的第一道屏障,其大部分血液供应来自肠道门静脉,而门静脉中富含细菌产物、环境毒素及食物抗原。受损的肝细胞会释放多种促炎细胞因子和趋化因子,激活肝脏内的驻留免疫细胞和浸润免疫细胞。肝脂肪变性与C反应蛋白水平升高(心血管不良结局的可靠预测指标)之间存在独立关联,这一事实凸显了肝脏在全身性炎症中所起的关键作用。此外,高盐摄入与非酒精性脂肪性肝病(NAFLD)及晚期肝纤维化风险增加独立相关。值得注意的是,作为心血管疾病的潜在独立危险因素,患有NAFLD个体发生10年心血管事件的风险更高。NAFLD可能通过引发全身性炎症、诱导胰岛素抵抗、增加氧化应激等方式促进高血压的发生,这与高盐的作用极为相似。然而,高盐诱导的NAFLD是否是心血管疾病发生发展的病理因素,目前尚不明确。
多种饮食和代谢因素(包括高糖、高脂以及盐介导的血管紧张素Ⅱ变化)导致的靶器官损伤,存在一种被称为“代谢记忆”的现象,即这些因素去除后,损伤仍持续存在。糖尿病肾病中代谢记忆机制的研究表明,炎症、氧化应激和表观遗传修饰是其主要成因。其中,活性氧(ROS)生成增加通过调控促炎基因启动子上的组蛋白表观遗传修饰,在糖尿病并发症中发挥核心作用,可导致低度慢性炎症状态。作为ROS的主要来源,线粒体功能障碍不仅会降低氧化呼吸链活性,导致肝脏脂肪沉积,还会触发先天免疫反应,从而促进NAFLD的发生。SIRT3(sirtuin 3)是抑制线粒体中ROS生成的主要去乙酰化酶,敲除小鼠SIRT3通过诱导多种线粒体蛋白的高乙酰化,导致脂肪肝和代谢综合征。但SIRT3是否参与盐诱导的肝脏炎症记忆,仍有待研究。
陆军军医大学陆军特色医学中心(大坪医院)祝之明主任医师课题组在Circulation期刊(IF=29.69)发表题为“Salt-Induced Hepatic Inflammatory Memory Contributes to Cardiovascular Damage Through Epigenetic Modulation of SIRT3”研究论文。该研究旨在探讨高盐饮食(HSD)诱导的肝脂肪变性是否是心血管损伤的主要原因,及其潜在机制。研究发现,肝脏中SIRT3表达降低是盐诱导的肝脏炎症和脂肪变性的重要介导因子。HSD通过表观遗传修饰机制抑制SIRT3的转录,导致肝脏炎症持续存在。值得注意的是,利用腺相关病毒8型(AAV8)载体在肝脏中过表达SIRT3有效缓解了小鼠持续性肝损伤的进展,从而对抗盐诱导的心血管损伤。综上所述,该研究结果表明,通过抑制肝脏慢性持续性炎症状态,有望为盐诱导的心血管损伤提供一种有效的治疗方法。
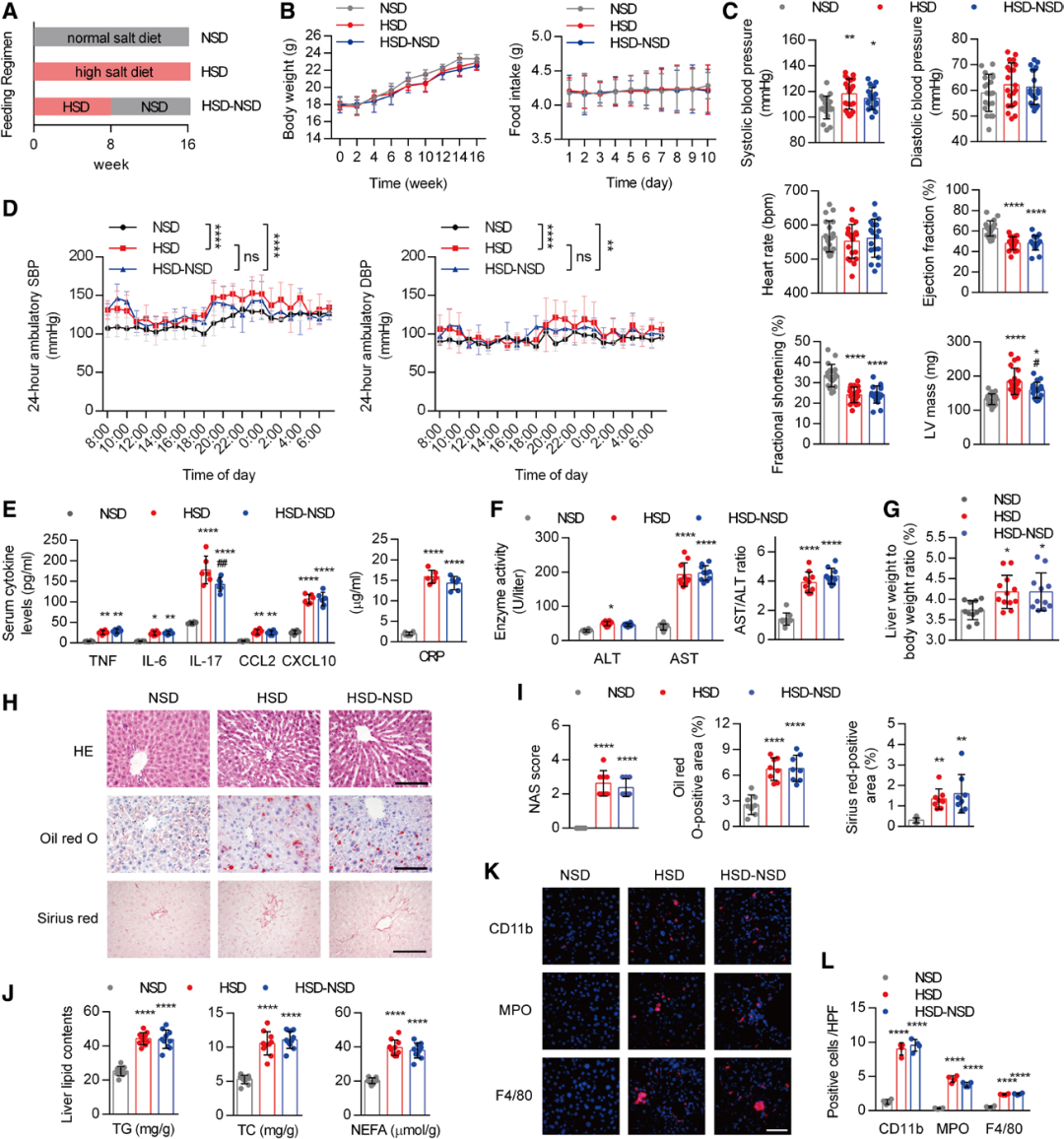
1.盐诱导的肝脏炎症和高血压在高盐摄入停止后仍持续存在
小鼠的干预过程和分组情况如图所示。三组小鼠的体重和食物摄入量无显著差异。高盐饮食(8%盐,HSD)显著升高收缩压,但不影响心率(HR);同时,心脏功能明显受损,表现为射血分数(EF)和短轴缩短率(FS)降低。此外,高盐摄入还显著提高了血清中促炎标志物(肿瘤坏死因子TNF、IL-6、IL-17、C-C基序趋化因子配体2/CCL2C-X-C基序趋化因子配体10/CXCL10及C反应蛋白/CRP)的水平。HSD使血清丙氨酸氨基转移酶(ALT)和天门冬氨酸氨基转移酶(AST)浓度(酶活)升高,并导致AST与ALT的比值上升。相应地,HSD喂养的小鼠肝重与体重的比值也更高。令人惊讶的是,停止高盐摄入后,全身性炎症和肝损伤均未恢复至正常水平,这表明存在持续性肝损伤。同样,盐诱导的高血压和心脏功能障碍在高盐摄入停止后仍较严重。此外,HSD还刺激了肝脏中的脂质蓄积、脂肪变性和纤维化,并使甘油三酯(TG)、总胆固醇(TC)和非酯化脂肪酸(NEFA)水平升高;HSD喂养的小鼠肝脏内有更多的中性粒细胞和巨噬细胞浸润。然而,改用正常盐饮食(0.4%盐,NSD)仍无法缓解这些肝脏病理变化。HSD导致炎症和纤维化相关基因的表达显著增加,而停止高盐摄入并未影响这些基因的表达。这些结果表明,高盐诱导的肝损伤具有“记忆”特征。
图1.盐诱导的炎症记忆存在于小鼠肝脏中,并促进心血管损伤
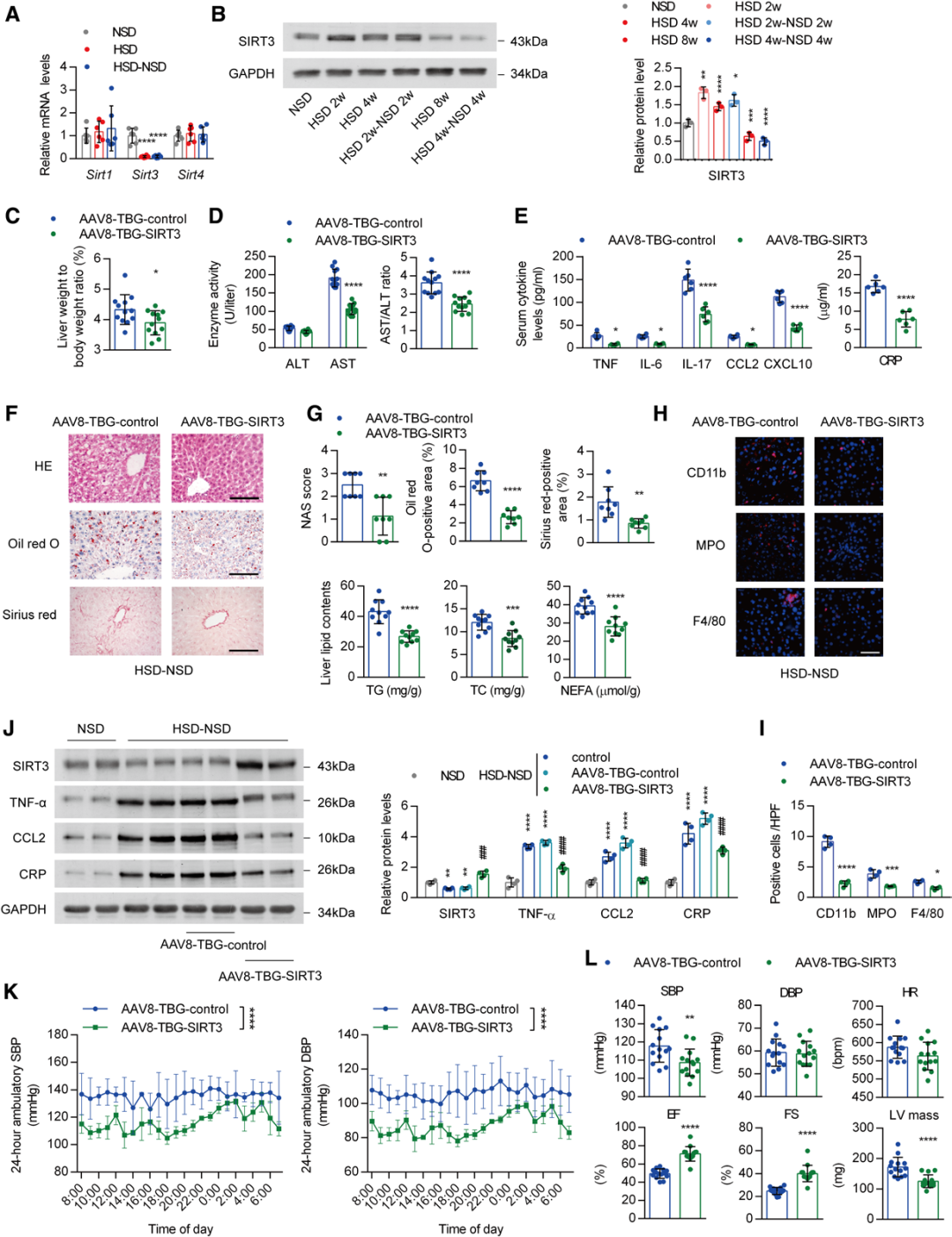
2.肝脏特异性过表达SIRT3可抑制盐诱导的肝脏炎症记忆及心血管损伤
线粒体功能障碍是导致肝脏炎症的重要因素。因此,研究人员检测了小鼠肝组织中的线粒体活性。结果显示,HSD显著降低了肝组织线粒体中复合体Ⅰ的氧化磷酸化活性以及复合体Ⅰ和Ⅳ的电子传递能力,且这些变化在停止高盐摄入后仍持续存在。由于SIRT3是一种关键的线粒体去乙酰化酶,研究者检测了其在肝脏中的表达水平。与细胞核中的SIRT1和线粒体中的SIRT4相比,HSD显著抑制了肝脏中SIRT3的表达,即使在停止高盐摄入后也是如此。短期HSD使SIRT3的表达升高,但长期HSD干预后其表达降低。因此,为探究SIRT3在记忆现象中的重要性,作者在HSD喂养的小鼠改用NSD之前,给其尾静脉注射了AAV8-TBG-SIRT3或对照病毒。在SIRT3敲除(SIRT3-/-)小鼠中验证了SIRT3在肝脏中表达的特异性。结果显示,除肝重与体重的比值降低外,注射AAV8-TBG-SIRT3的小鼠AST水平和促炎细胞因子水平也显著降低。此外,在停止高盐摄入后,SIRT3过表达显著减轻了肝细胞脂肪变性、脂质蓄积、纤维化以及炎症细胞浸润。SIRT3过表达还显著抑制了促炎基因的表达。因此,肝脏中的记忆现象可能与SIRT3的持续低表达有关。
出乎意料的是,停止高盐摄入后,AAV介导的肝脏中SIRT3过表达不仅显著降低了盐诱导的高血压,还拮抗了心脏功能障碍,这表明肝脏中SIRT3的低表达也是盐诱导心血管损伤的重要原因。
图2.肝脏中过表达SIRT3可拮抗停止高盐摄入后持续的炎症反应和心血管损伤
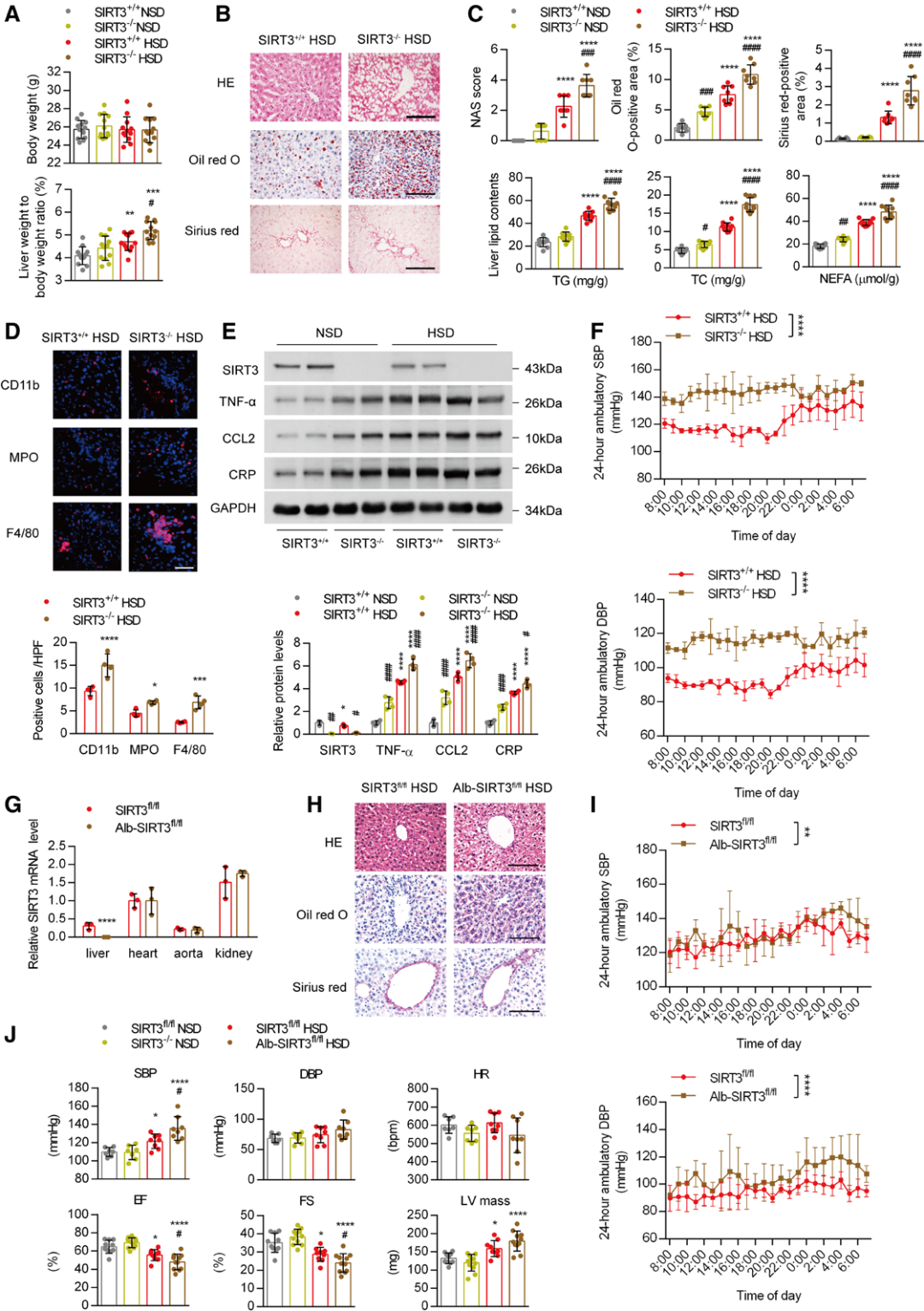
3.肝脏中SIRT3的持续减少导致盐诱导的炎症记忆
接下来,研究人员使用SIRT3-/-小鼠模拟SIRT3表达降低的状态。通过RNA测序,观察到HSD和SIRT3敲除共同上调的基因富集于免疫系统,包括TNF、CCL2等。在NSD喂养状态下,SIRT3敲除对肝重与体重的比值无影响,但进一步加剧HSD诱导的该指标升高。在SIRT3-/-小鼠中,HSD诱导的肝损伤、组织学改变及炎症细胞浸润也更为严重。此外,SIRT3敲除直接增加了促炎基因的表达,并进一步加剧了HSD对这些细胞因子的促进作用。相应地,SIRT3敲除还加重了盐诱导的高血压和心脏功能障碍。在NSD和HSD组中,SIRT3敲除共同上调的基因仍富集于免疫系统,这表明SIRT3在肝脏炎症中具有直接作用。
为进一步明确SIRT3在肝脏中的作用,研究者利用Cre-loxP系统构建了肝脏特异性SIRT3敲除小鼠,并给予HSD喂养。与预期一致,与同窝的SIRT3flox/flox小鼠相比,肝脏中SIRT3敲除的小鼠也出现了更严重的肝脂肪变性和纤维化,同时伴有血压升高和心脏功能下降。
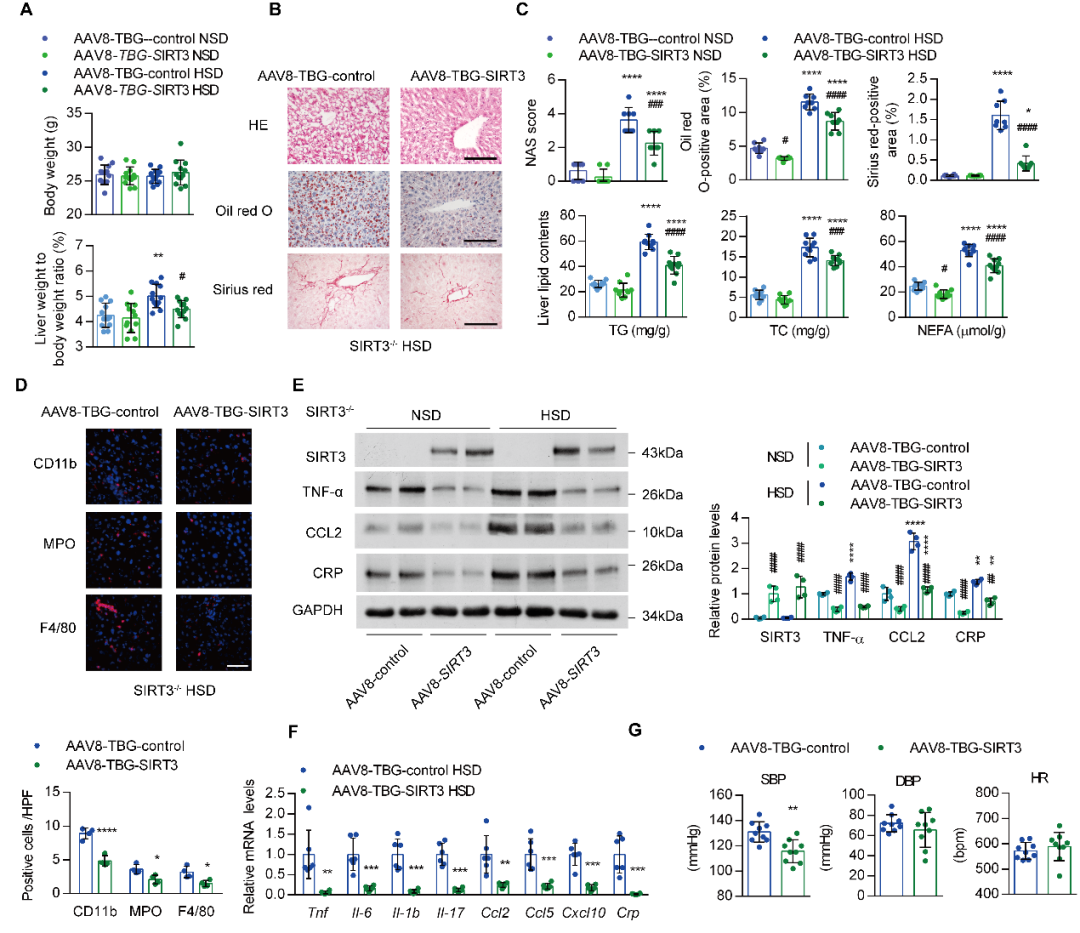
随后,作者通过给SIRT3-/-小鼠注射AAV8-TBG-SIRT3,评估了SIRT3对HSD诱导的肝脏炎症的保护作用。结果显示,恢复SIRT3的表达显著降低了HSD喂养的SIRT3-/-小鼠的肝重与体重比值。与注射对照载体的小鼠相比,注射AAV8-TBG-SIRT3的SIRT3-/-小鼠的肝脂肪变性、脂质蓄积、纤维化及炎症细胞浸润均显著减轻。基因表达分析还表明,在SIRT3-/-小鼠中过表达SIRT3不仅直接降低了促炎细胞因子的高表达水平,还阻断了HSD对其的促进作用。肝脏中SIRT3的过表达也降低了SIRT3-/-小鼠中盐诱导的收缩压升高。
图3.肝脏中敲除SIRT3会加剧高盐诱导的肝脏炎症和心血管损伤
图4.SIRT3-/-小鼠中过表达SIRT3可减轻HSD诱导的肝脏炎症
4.HSD通过表观遗传方式降低SIRT3的表达
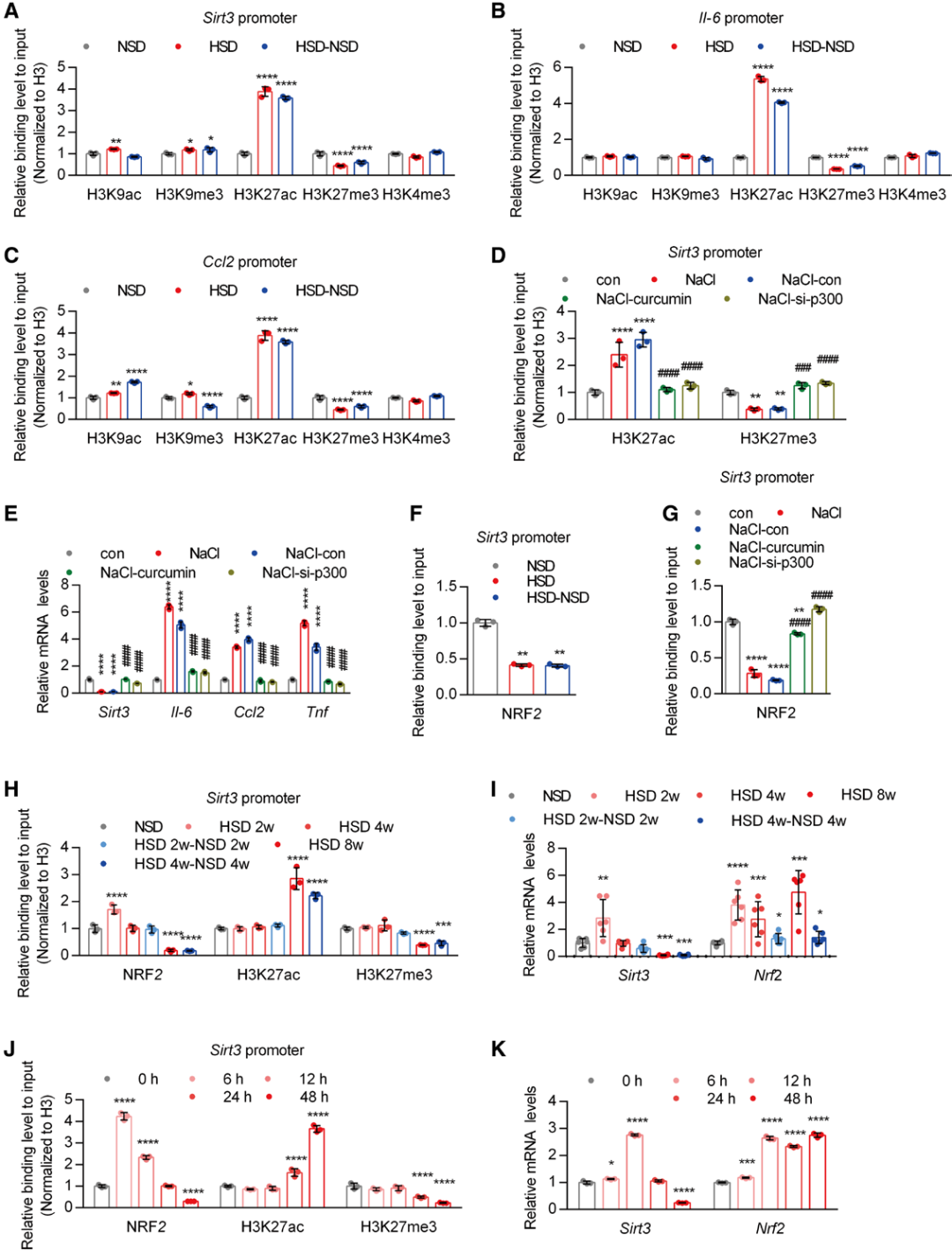
由于DNA甲基化在抑制基因表达中发挥重要作用,作者首先探究了与Sirt3基因相关的DNA甲基化状态,以解释盐负荷下SIRT3持续低表达的原因。尽管HSD确实增加了肝组织的整体DNA甲基化水平,但从全基因组亚硫酸氢盐测序(WGBS)数据中,未观察到Sirt3基因启动子区域CpG岛的DNA甲基化发生任何变化。因此,转而研究组蛋白表观遗传修饰,发现HSD显著提高了肝脏中p300的表达水平——p300是组蛋白3赖氨酸27(H3K27)的乙酰转移酶,但其甲基转移酶(Ezh2)的表达未受影响。不过,p300的乙酰转移酶活性并未改变。通过染色质免疫沉淀(ChIP)测序,研究人员确定Sirt3基因启动子区域存在乙酰化H3K27(H3K27ac)峰,且高盐负荷后H3K27ac的富集程度增加。ChIP结果还显示,HSD喂养的小鼠肝组织中,H3K27ac与SIRT3启动子的结合显著增加,同时该位点的三甲基化(H3K27me3)水平降低。这些组蛋白修饰的变化在高盐去除后仍持续存在。此外,探究H3K27ac水平升高是否对高盐的基因表达调控作用至关重要。结果显示,小鼠原代肝细胞停止高盐负荷后,敲低或抑制p300(姜黄素)显著消除了H3K27的高乙酰化,并降低促炎基因的表达水平。同时,Sirt3的低表达也恢复至正常水平。
然而,为何H3K27ac对SIRT3表达的影响与对促炎因子的影响相反?作者重新分析了Sirt3基因的启动子区域,发现其中存在NRF2(核因子E2相关因子2)的结合位点——NRF2是SIRT3的正向调控因子。实验发现,HSD显著抑制了肝组织中NRF2与Sirt3启动子区域的结合,且这种抑制在改用NSD后仍持续存在。重要的是,敲低或抑制p300恢复了高盐导致的NRF2与Sirt3启动子结合水平的降低,这表明H3K27ac水平升高可能降低了NRF2的结合能力。此外,2周的HSD显著刺激了小鼠肝脏中NRF2与SIRT3启动子的结合水平,同时伴随SIRT3的短暂高表达。但随着时间推移,NRF2的结合减少,而H3K27ac逐渐增加,同时SIRT3的表达降低。此外,NRF2的表达依赖于HSD的存在,因为在高盐去除后其表达恢复正常。SIRT3的表达先升高后下降,几乎与其启动子上NRF2的结合水平一致。因此,这些结果表明,H3K27ac对NRF2结合的抑制作用,是高盐去除后SIRT3表达持续降低的主要原因。
图5.H3K27ac通过抑制SIRT3启动子上NRF2的结合,降低SIRT3的表达
5.NRF2介导表观遗传修饰对SIRT3表达的调控作用
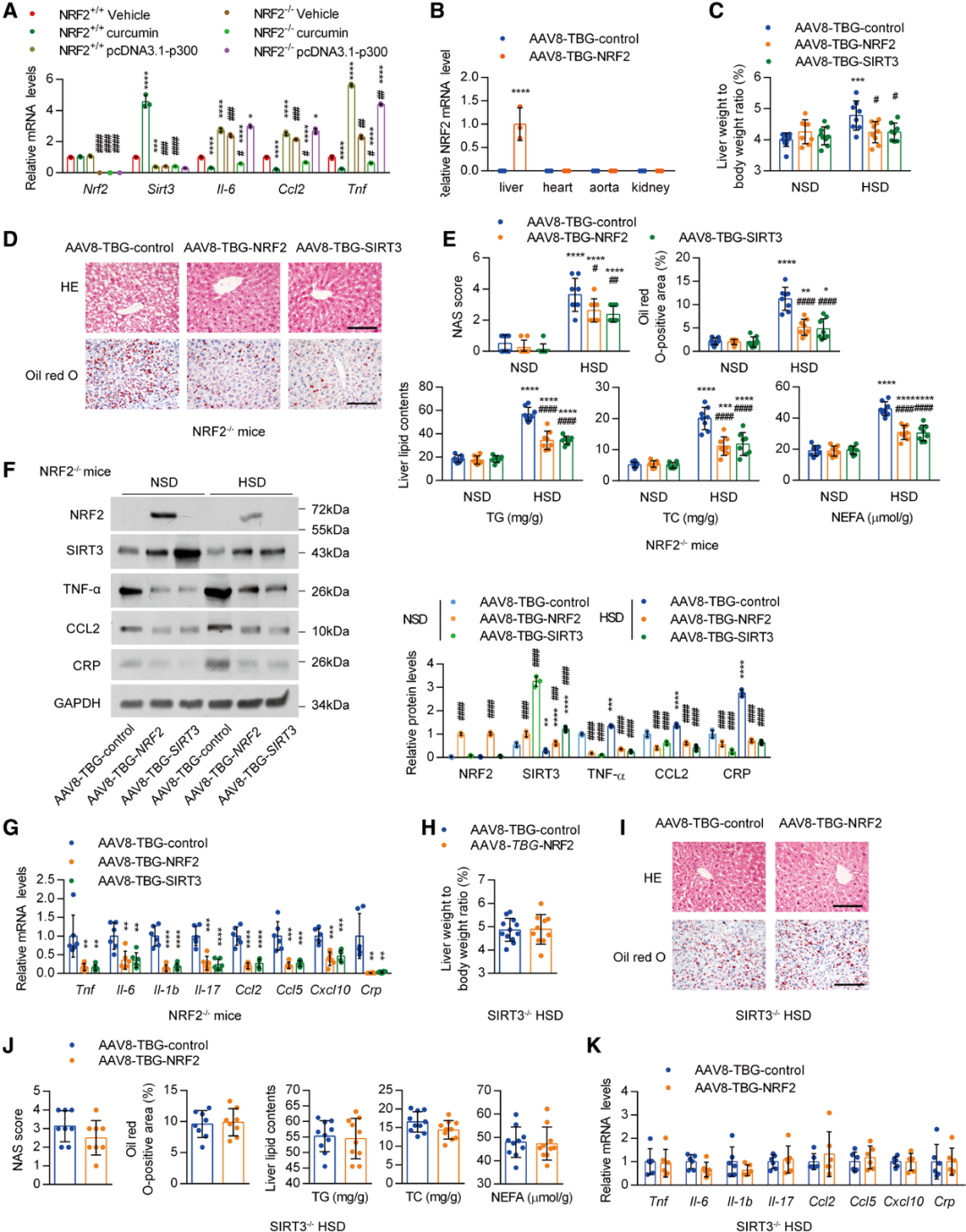
为明确NRF2在组蛋白乙酰化对SIRT3表达的抑制效应中是否不可或缺,作者用p300抑制剂姜黄素或p300过表达质粒处理来自NRF2+/+和NRF2-/-小鼠的原代肝细胞。结果显示,姜黄素直接刺激SIRT3表达,同时降低炎症分子的表达;而p300过表达则显著降低SIRT3的表达,并升高炎症分子的表达。此外,敲除NRF2直接降低了SIRT3的表达水平,并消除姜黄素或p300过表达质粒的调控作用。然而,这些干预对促炎细胞因子编码基因的调控作用并未受到NRF2敲除的影响。这些结果表明,NRF2与SIRT3启动子结合的受限,是高盐诱导的组蛋白表观遗传修饰对SIRT3表达产生抑制效应的关键因素。
接下来,为验证NRF2对肝脏SIRT3表达是否不可或缺,研究者给NRF2-/-小鼠喂食HSD 16周。结果显示,NRF2缺失对肝重与体重的比值无明显影响,但进一步加剧HSD对该比值的促进作用。在高盐负荷下,NRF2-/-小鼠还表现出更严重的肝脂肪变性。相应地,HSD喂养的NRF2-/-小鼠肝脏中,促炎细胞因子的表达水平显著升高,同时SIRT3的表达降低。
因此,为证实SIRT3的抑制是否介导了NRF2敲除加剧肝脏炎症的作用,研究人员给NRF2-/-小鼠分别注射了AAV8-TBG-SIRT3、AAV8-TBG-NRF2和对照载体。AAV8-TBG-NRF2在肝脏中的特异性也得到了验证。与AAV8-TBG-NRF2类似,恢复肝脏中SIRT3的表达也显著阻断了NRF2敲除对HSD诱导的肝损伤的促进作用。此外,SIRT3过表达还改善了HSD喂养的NRF2-/-小鼠肝脏中促炎细胞因子编码基因的高表达。相反,在HSD喂养的SIRT3-/-小鼠肝脏中过表达NRF2,发现AAV8-TBG-NRF2无法减轻小鼠的肝脂肪变性和脂质蓄积。两组小鼠的炎症基因表达也无差异。这些结果表明,作为NRF2的下游靶标,SIRT3表达的抑制是NRF2敲除促进肝脏炎症的主要原因,且NRF2的肝脏保护作用需要SIRT3的参与。
图6.肝脏过表达SIRT3可减轻NRF2-/-小鼠的持续性炎症
6.二甲双胍治疗可阻断盐诱导的肝脏炎症记忆及心血管损伤
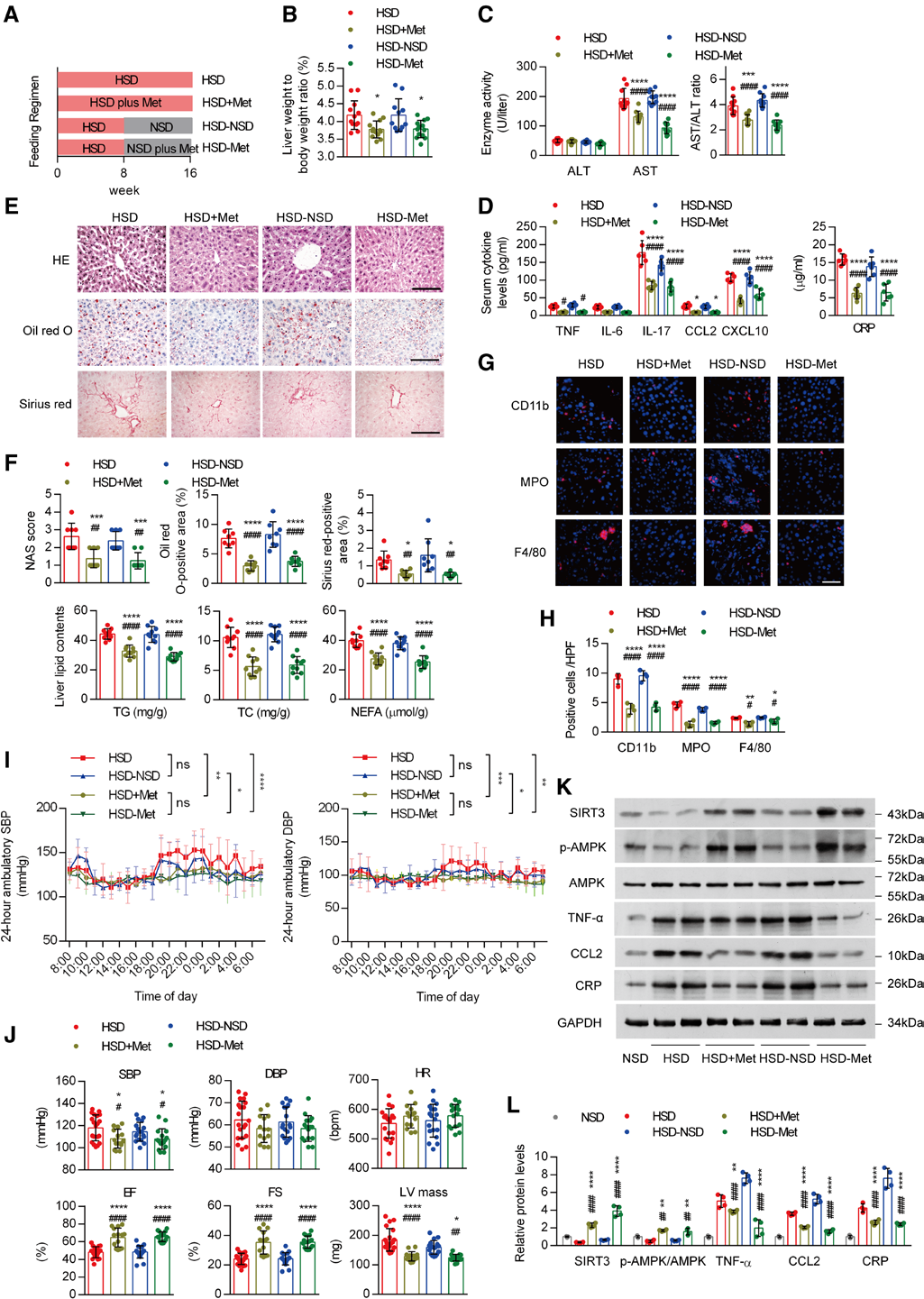
接下来,作者试图探究何种干预措施可对抗盐诱导的炎症记忆。ChIP测序结果显示,SIRT3敲除主要影响与AMPK(腺苷酸活化蛋白激酶)和胰岛素信号通路相关的基因。由于二甲双胍是已知的AMPK激活剂,且能改善高糖或高脂饮食(HFD)诱导的持续性靶器官损伤。研究人员发现,二甲双胍干预后,降低了小鼠的肝重与体重比值,血清AST/ALT比值和促炎标志物水平也显著降低;减轻了HSD诱导的肝损伤和炎症,还完全阻断了盐记忆现象;HSD引起的持续性高血压和心脏功能障碍显著改善。基因表达分析显示,二甲双胍显著降低了HSD喂养小鼠中与炎症和纤维化相关基因的表达升高,并阻断了高盐去除后这些基因的高表达,同时伴随SIRT3表达升高和AMPK激活。二甲双胍治疗还改善了HSD喂养小鼠肝脏的线粒体功能。ChIP结果表明,二甲双胍几乎完全消除了HSD诱导的H3K27高乙酰化,显著提高了NRF2与SIRT3启动子的结合,同时降低了p300基因的表达水平。此外,二甲双胍还阻断了高盐去除后这些指标的持续状态。这些结果表明,二甲双胍对盐诱导的肝脏炎症记忆具有强效抑制作用。
作者进一步利用AMPKα2-/-小鼠,发现敲除AMPKα2不仅阻断了二甲双胍对HSD诱导的肝脂肪变性的预防作用,还抑制了二甲双胍在高盐去除后对这些特征的改善作用。在AMPKα2-/-小鼠中,二甲双胍对SIRT3表达的促进作用消失,同时伴随AMPK失活。这些结果表明,AMPK是二甲双胍发挥预防作用所必需的,且SIRT3的激活也依赖于AMPK。
图7.二甲双胍抑制盐诱导的炎症记忆和心血管损伤的发生
总结
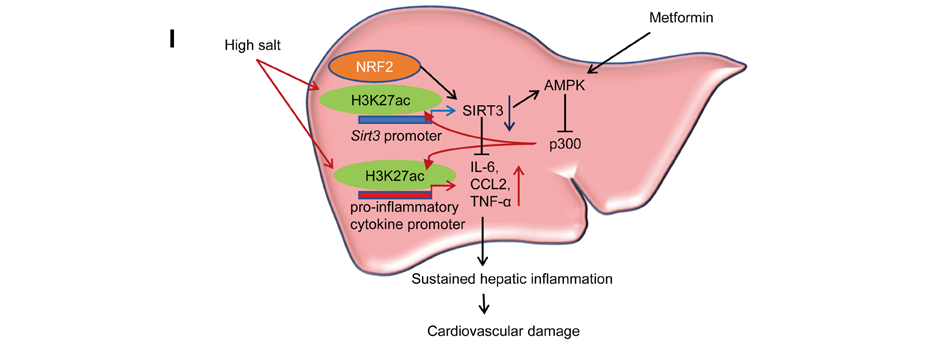
本研究指出,HSD诱导的肝脂肪变性和炎症会表现出炎症记忆现象,并促成心血管损伤,其中HSD通过表观遗传修饰降低肝脏SIRT3的表达,这一过程起到了关键作用。SIRT3表达水平持续偏低是由于H3K27的过度乙酰化抑制了NRF2与其启动子的结合。此外,二甲双胍通过激活AMPK,对盐诱导的肝脏炎症具有保护作用,这提示在HSD人群中,二甲双胍可能成为一种颇具前景的预防心血管损伤的干预手段。
图8.HSD通过肝脏炎症记忆促成心血管损伤模式图
该研究获得国家自然科学基金(项目编号:31430042、31871199、82022006、81721001和81873657)、国家重点研发计划(项目编号:2018YFA0800601)等项目的资助。
本文使用的病毒产品,列表如下:
上述病毒产品我司均可提供,欢迎咨询!

了解产品及服务
请扫码添加客服微信:BrainVTA2020

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK