2025-05-26 阅读量:3788
非酒精性脂肪肝病(NAFLD)是全球最常见的慢性肝脏疾病(患病率约为25%),是一系列肝脏疾病的总称,包括非酒精性脂肪肝(NAFL,又称单纯性脂肪肝,多数患者无症状)和非酒精性脂肪性肝炎(NASH)两种主要病理类型。肝脏脂肪堆积可使肝脏对损伤因素(如氧化应激、炎症因子、药物毒性等)的敏感性增加,从而更易发生肝损伤,进而发展为NASH。NASH的特征是肝脏存在不同程度的炎症和纤维化。中心性肥胖、糖尿病(DM)和胰岛素抵抗(IR)是肝脂肪变性发展的重要危险因素。尽管导致NASH进展的炎症和纤维化机制尚未明确,但新证据表明肝脏脂毒性是理解NASH发病机制的关键。肝脏脂毒性指某些脂质分子在肝细胞内的积累可直接产生细胞毒性,或通过促炎、促纤维化作用引发病变。虽然甘油三酯(TG,NASH和脂肪变性时肝脏主要脂质成分)被认为是安全的储存性脂质,但少量具有脂毒性的游离胆固醇(FC)和游离脂肪酸(FFA)及其衍生物可能对NASH发展产生显著负面影响。
研究表明,胆固醇稳态失调是NAFLD的重要特征,表现为胆固醇合成和摄取增加,而外排和排泄相对减少,导致肝脏中胆固醇水平升高。肝细胞获取胆固醇的主要途径包括:HMGCR(3-羟基-3-甲基戊二酰辅酶A还原酶,限速酶)介导的内源性合成(甲羟戊酸途径)、低密度脂蛋白受体(LDLR)介导的内吞作用摄取低密度脂蛋白颗粒,以及通过SR-BI(清道夫受体B类成员1,编码基因为SCARB1)直接摄取循环中的高密度脂蛋白胆固醇酯(CEs)。而肝细胞清除胆固醇的主要途径包括:转化为胆汁酸[经典胆汁酸合成(BAS)途径(中性途径)限速酶为胆固醇-7α羟化酶(即细胞色素P450家族7亚家族A成员1/CYP7A1)、替代BAS途径(酸性途径)关键酶为甾醇-27-羟化酶(即细胞色素P450家族27亚家族A成员1/CYP27A1)],再经胆盐输出泵(BSEP)将胆汁酸排泄到胆汁中;通过ABCG5/G8(ATP结合组件亚家族G成员5及成员8,转运蛋白)将胆固醇排泄到胆汁中;通过ATP结合组件亚家族A成员1(ABCA1,转运蛋白)转运FC,将胆固醇流出到循环载脂蛋白AI和新生高密度脂蛋白颗粒上。此外,ACAT2(乙酰-CoA乙酰转移酶2)可将FC酯化为CEs,并促进CEs结合到极低密度脂蛋白颗粒中,然后分泌到血液中。例如,肝脏锌指蛋白36样蛋白1(ZFP36L1)缺失的小鼠可通过CYP7A1介导的BAS增强,从而抵抗饮食诱导的肥胖和脂肪变性;肝细胞中过表达核因子-4α可通过协调调控CYP7A1和CYP8B1[细胞色素P450家族8亚家族B成员1,BAS经典途径中决定胆酸(CA)生成的关键酶]介导的胆汁酸信号通路,减轻高脂/高胆固醇/高果糖(HFCF)饮食诱导的脂肪性肝炎。因此,通过干预这些通路减少肝脏FC(具有脂毒性的分子)的蓄积可能是改善NAFLD的可行策略。
G蛋白偶联受体(GPCRs)能够感知多种细胞外分子,激活细胞内信号传导并引发多种细胞反应。研究表明,GPCRs在NAFLD中具有重要作用,例如GPR120、GPR132、GPR55和GPR91等受体通过结合胆汁酸和FFA参与疾病进程。其中,胆汁酸激活的膜结合型GPCR——武田G蛋白偶联受体5(TGR5)已被证实可刺激能量代谢,保护肝脏和肠道免受炎症及脂肪变性影响,并改善胰岛素敏感性。另一个新近发现的胆汁酸激活GPCR——鞘氨醇-1-磷酸受体2(sphingosine-1-phosphate receptor 2,S1PR2),可能通过协同激活细胞外信号调节蛋白激酶1/2(ERK1/2)、蛋白激酶B(PKB)和法尼醇X受体(FXR),在调节肝脏葡萄糖、胆汁酸及脂质代谢中发挥重要作用。然而,目前尚未明确介导胆固醇向胆汁酸转化的特异性GPCR分子。
GPR35是一种研究较少的“孤儿”受体。已有证据表明GPR35参与肥胖和DM的病理过程。在炎症和代谢应激的实验模型及生理条件下,GPR35的内源性激动剂犬尿喹.啉酸(Kyna)的循环水平显著升高。在肥胖和2型糖尿病(T2DM)中,脂肪组织中的GPR35可介导Kyna的作用,包括减少体重增加和增强脂肪组织的能量消耗。值得注意的是,GPR35基因的编码区及基因间区遗传学异常与高血压、动脉粥样硬化斑块形成及T2DM等代谢性疾病相关。这些发现强烈支持GPR35信号在炎症和代谢应激中的关键作用,但其分子机制及靶向的毒性脂质尚未明确,尤其在肥胖相关NAFLD中GPR35在肝细胞内的具体功能仍属未知。
近日,安徽医科大学王学富教授和王华教授课题组合作,在药学领域顶刊《药学学报(Acta Pharmaceutica Sinica B)》(中科院一区,IF=14.9)上发表题为《G蛋白偶联受体35通过重编肝脏胆固醇稳态来减轻非酒精性脂肪性肝炎(G protein-coupled receptor 35 attenuates nonalcoholic steatohepatitis by reprogramming cholesterol homeostasis in hepatocytes)》的学术论文。本研究发现,肝细胞GPR35通过调节肝脏胆固醇稳态来缓解NASH。具体而言,肝细胞中过表达GPR35可抵抗HFCF饮食诱导的脂肪性肝炎,而GPR35缺失则产生相反效果。给予GPR35激动剂Kyna可抑制HFCF饮食诱导的小鼠脂肪性肝炎。Kyna/GPR35通过ERK1/2信号通路诱导StAR相关脂质转移蛋白4(STARD4)表达,最终促进肝脏胆固醇酯化和BAS。过表达STARD4可上调BAS限速酶CYP7A1和CYP8B1的表达,促进胆固醇向胆汁酸转化。在肝细胞特异性STARD4敲低小鼠中,GPR35过表达的保护作用消失。肝细胞过表达STARD4可逆转GPR35缺失对HFCF饮食诱导脂肪性肝炎的加重效应。这些发现表明GPR35-STARD4轴是治疗NAFLD的潜在靶点。
1.全身性GPR35敲除加剧HFCF饮食诱导的脂肪性肝炎
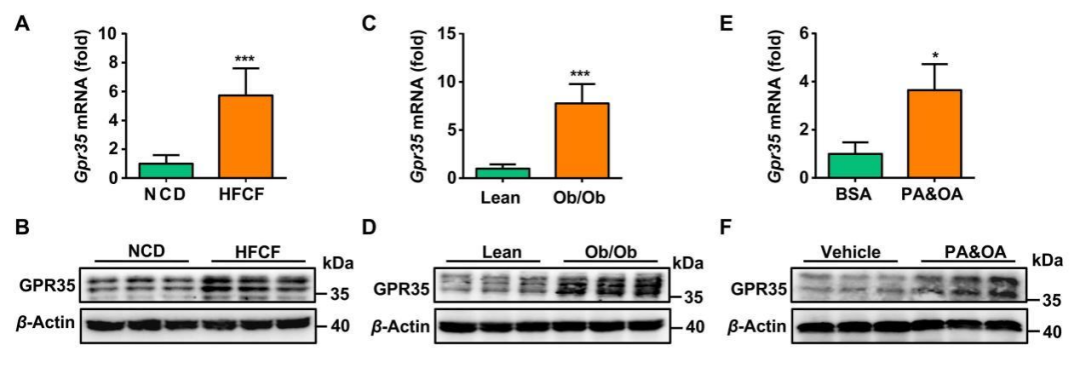
为探究GPR35在肝脏代谢中的作用,研究人员检测了NAFLD小鼠肝脏中GPR35的表达水平。8周龄C57BL/6J小鼠分别接受HFCF饮食或正常对照饮食(NCD)喂养16周。结果显示,与对照组相比,HFCF饮食小鼠肝脏中GPR35的蛋白及mRNA表达显著上调。此外,ob/ob肥胖小鼠肝脏的GPR35表达也较瘦型小鼠明显升高。为进一步验证这一现象是否发生于肝细胞,研究者从野生型(WT)小鼠中分离原代肝细胞,并在体外使用棕榈酸&油酸(PA&OA,肝细胞脂质蓄积和IR诱导剂)刺激。相比于对照组,PA&OA处理显著增加了肝细胞中GPR35的表达。
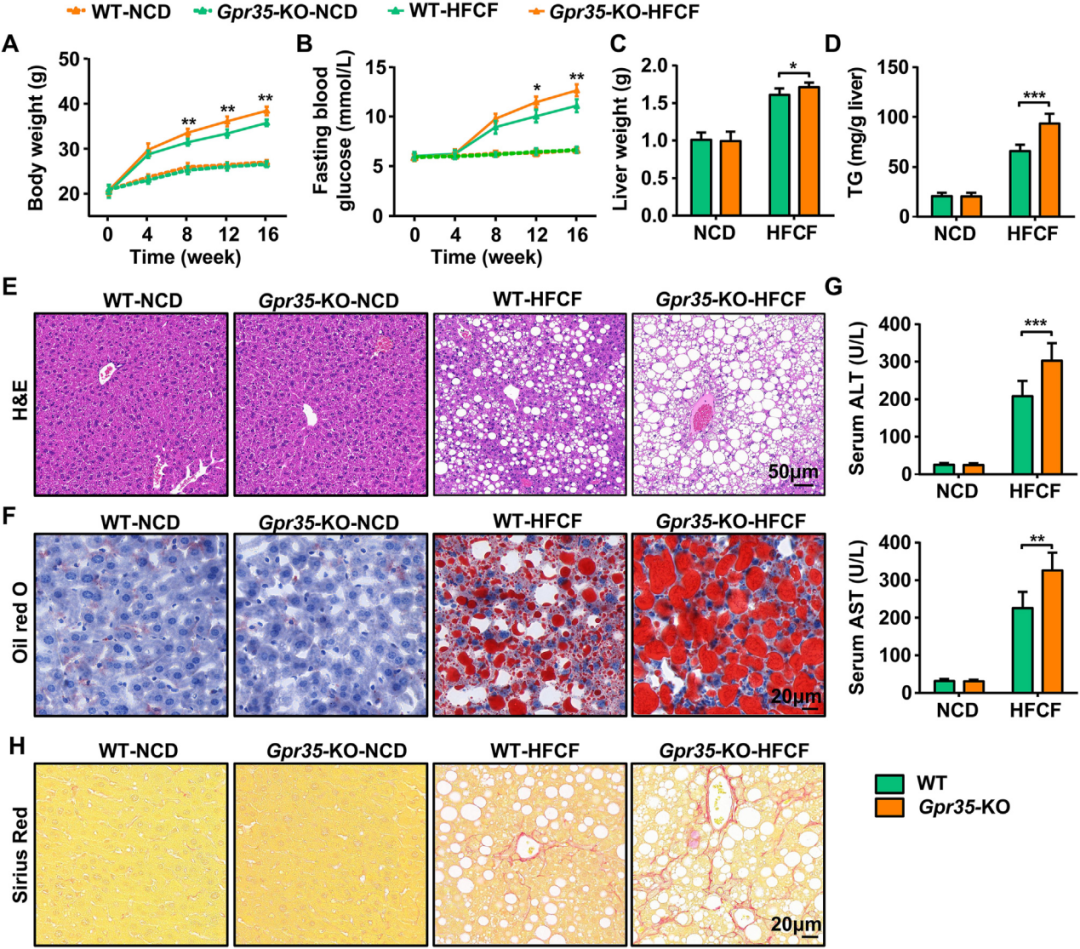
随后,作者研究了GPR35在NAFLD中的功能。将全身性GPR35敲除(Gpr35-KO)小鼠与WT小鼠分别接受NCD或HFCF饮食喂养16周。结果显示,在HFCF饮食组中,Gpr35-KO小鼠的体重和空腹血糖(FBG)水平均显著高于WT小鼠;肝脏重量和TG含量也显著高于WT小鼠。油红O和苏木.精-伊红(H&E)染色显示,Gpr35-KO小鼠肝脏的脂肪变性及脂质蓄积更为严重。与脂肪变性程度一致,GPR35全身性敲除还导致血浆ALT(丙氨酸氨基转移酶)和AST(天冬氨酸氨基转移酶)[肝功能指标]水平升高,并诱发更显著的肝纤维化(天狼星红染色)。综上,GPR35缺失加剧了饮食诱导的NASH进展。
图1.肝细胞中GPR35表达在NAFLD中显著增加
图2.全身性敲除GPR35加剧了HFCF饮食诱导的脂肪性肝炎
2.肝细胞GPR35表达缺失加剧HFCF饮食诱导的脂肪性肝炎及IR
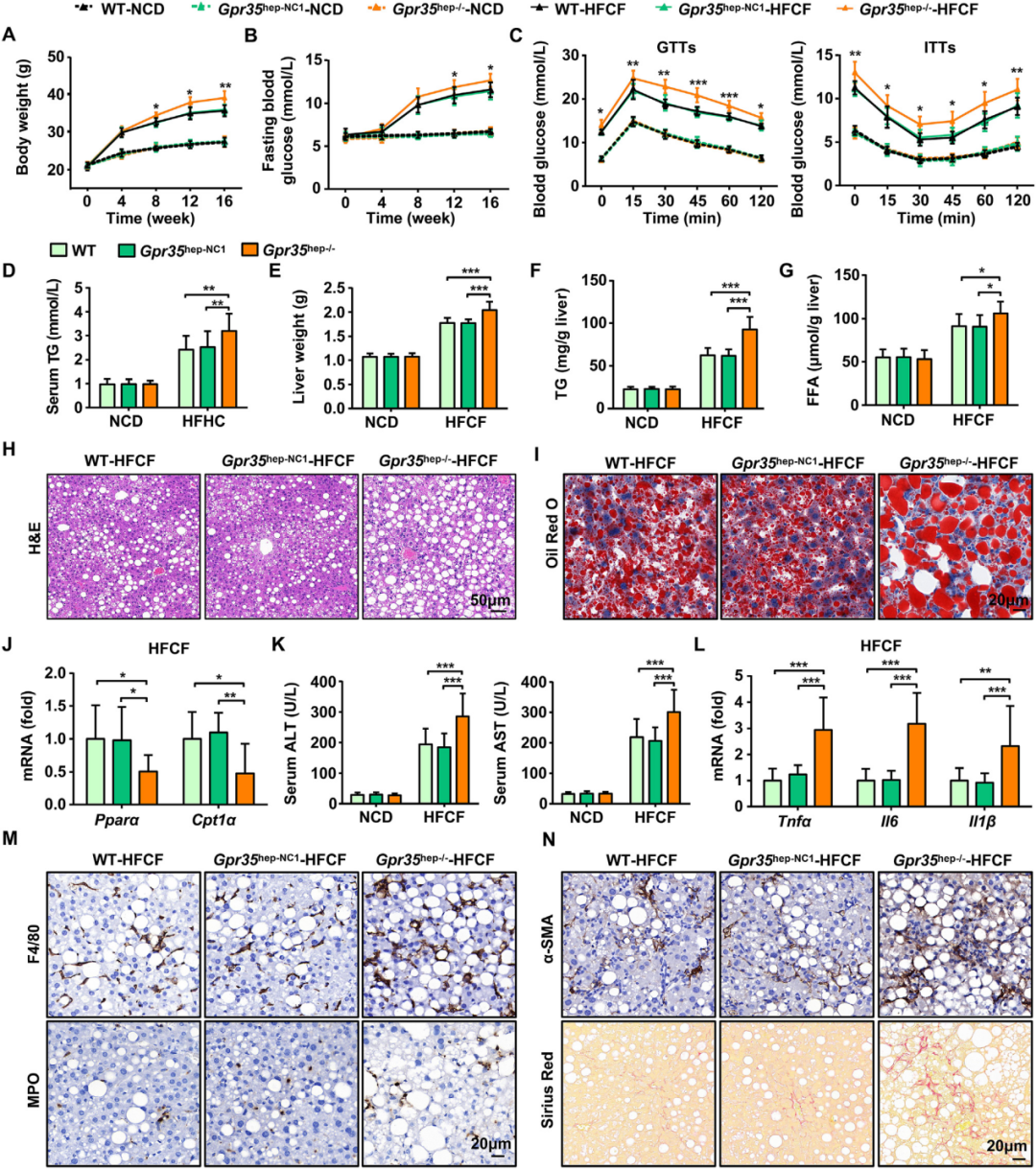
为明确肝细胞GPR35表达对NASH及IR的体内调控作用,研究人员通过静脉注射携带靶向Gpr35的sgRNA的腺相关病毒8型载体[AAV8-TBG-SaCas9-2A-EGFP-U6-sgRNA(Gpr35)],构建了肝细胞特异性GPR35敲除小鼠(Gpr35hep−/−)。随后,将Gpr35hep−/−小鼠、对照小鼠[Gpr35hep-NC1,注射AAV8-TBG-SaCas9-2A-EGFP-U6-sgRNA(NC)]及WT小鼠分别接受HFCF饮食或NCD喂养16周。结果显示,在NCD喂养条件下,肝细胞GPR35缺失对小鼠体重、IR及肝脏脂肪变性无显著影响。
在HFCF饮食组中,WT小鼠的体重和FBG水平显著升高,而Gpr35hep−/−小鼠的体重及FBG水平较Gpr35hep-NC1和WT小鼠(统称对照组)进一步增加。通过葡萄糖耐量试验(GTT)和胰岛素耐量试验(ITT)发现,与对照组比较,Gpr35hep−/−小鼠的葡萄糖耐受性下降且胰岛素敏感性受损;血清TG水平也显著高于对照组。这些数据表明,肝细胞GPR35表达缺失促进了HFCF饮食诱导的肥胖及IR。
此外,与对照组相比,Gpr35hep−/−小鼠的肝脏重量显著增加,肝脏TG和FFA水平也显著升高。H&E和油红O染色显示,肝细胞GPR35缺失加剧了肝脏脂肪变性。尽管与脂肪从头合成(DNL)相关的基因(Srebf1:编码固醇调节元件结合蛋白1c/Srebp-1c;Acaca/Acc1:编码乙酰辅酶A羧化酶α;Fasn/FAS:编码脂肪酸合酶)表达水平未显著变化,但脂肪酸氧化(FAO)相关基因(Cpt1a:编码肉碱棕榈酰转移酶1A;Ppara:编码过氧化物酶体增殖物激活受体α)的mRNA表达因GPR35缺失而受到抑制。
进一步分析发现,肝细胞GPR35缺失加剧了HFCF饮食诱导的肝损伤、炎症及纤维化。具体而言,HFCF饮食组中,Gpr35hep−/−小鼠的血清ALT和AST水平显著高于对照组。此外,Gpr35hep−/−小鼠肝脏中促炎细胞因子(肿瘤坏死因子-α/TNF-α、白介素-6/IL-6、白介素-1β/IL-1β)的mRNA表达显著上调。免疫组化染色显示,Gpr35hep−/−小鼠肝脏中巨噬细胞标志物F4/80及中性粒细胞标志物髓过氧化物酶(MPO)的阳性信号增强,提示炎症细胞浸润增加。天狼星红染色及免疫组化染色显示,Gpr35hep−/−小鼠的肝脏α-SMA水平升高,纤维化程度加重。综上,肝细胞GPR35表达缺失显著恶化了HFCF饮食诱导的脂肪性肝炎及IR。
图3.肝细胞中GPR35的缺失加剧了HFCF饮食诱导的脂肪性肝炎和IR
3.肝细胞GPR35过表达抵抗HFCF饮食诱导的脂肪性肝炎和IR
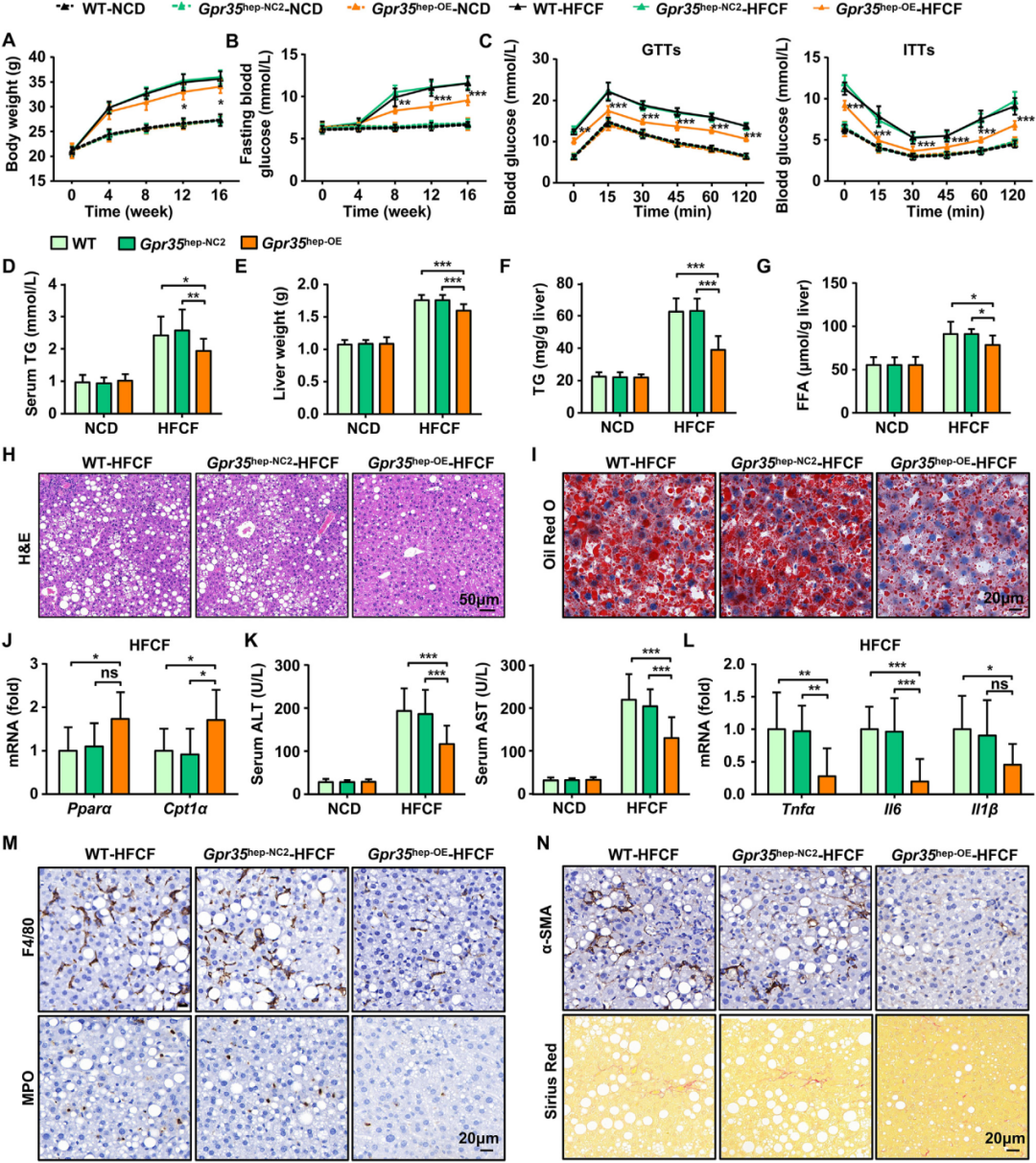
除了采用功能缺失性研究策略外,作者通过向WT小鼠注射AAV8-TBG-MCS-Gpr35-EGFP-3Flag-SV40 PolyA,构建了肝细胞特异性过表达GPR35的小鼠模型(Gpr35hep-OE小鼠),以探究肝细胞中GPR35过表达是否能改善HFCF饮食诱导的NAFLD。将Gpr35hep-OE、对照(Gpr35hep-NC2,注射AAV8-TBG-MCS-EGFP-3Flag-SV40 PolyA)和WT小鼠分别饲喂HFCF饮食或NCD 16周。结果显示,在NCD喂养条件下,肝细胞GPR35过表达对小鼠体重、IR或肝脏脂肪变性均无显著影响。然而,与Gpr35hep-NC2或WT小鼠相比,HFCF饮食诱导的Gpr35hep-OE小鼠体重增幅和FBG水平升高幅度更小。GTT和ITT表明,HFCF饮食喂养的Gpr35hep-OE小鼠的葡萄糖耐受性和胰岛素敏感性显著增强。此外,这些小鼠的血清TG水平也显著降低,提示肝细胞GPR35过表达减轻了HFCF饮食诱导的肥胖和IR。
在HFCF饮食干预下,与Gpr35hep-NC2和WT小鼠相比,Gpr35hep-OE小鼠的肝脏重量、TG含量及FFA含量均显著下降。与上述结果一致,肝细胞GPR35过表达上调了FAO相关基因的mRNA表达,但对DNL相关基因无显著影响。通过肝脏切片染色(H&E和油红O染色)进一步证实,HFCF饮食喂养的Gpr35hep-OE小鼠肝脏脂质沉积和脂肪变性显著减轻。
此外,HFCF饮食诱导的肝损伤指标(血清ALT和AST水平)在Gpr35hep-OE小鼠中显著降低。肝细胞GPR35过表达还抑制了HFCF饮食诱导的肝脏促炎细胞因子表达及炎症细胞浸润。更重要的是,GPR35过表达缓解了HFCF饮食诱导的肝纤维化。综上所述,这些结果表明肝细胞中GPR35的过表达对HFCF饮食诱导的小鼠NAFLD具有保护作用。
图4.肝细胞中过表达GPR35的小鼠对HFCF饮食诱导的脂肪性肝炎和IR具有抗性
4.GPR35通过胆固醇酯化和肝脏BAS调控胆固醇稳态
越来越多的证据表明,胆固醇稳态紊乱和肝脏FC蓄积与NASH的发病机制密切相关。在NAFLD患者中,NASH和肝纤维化的进展与肝脏FC蓄积呈平行关系。实验性诱导肝脏FC蓄积会加重脂肪性肝炎和纤维化,而纠正肝脏FC超载可改善NASH的疾病严重程度。
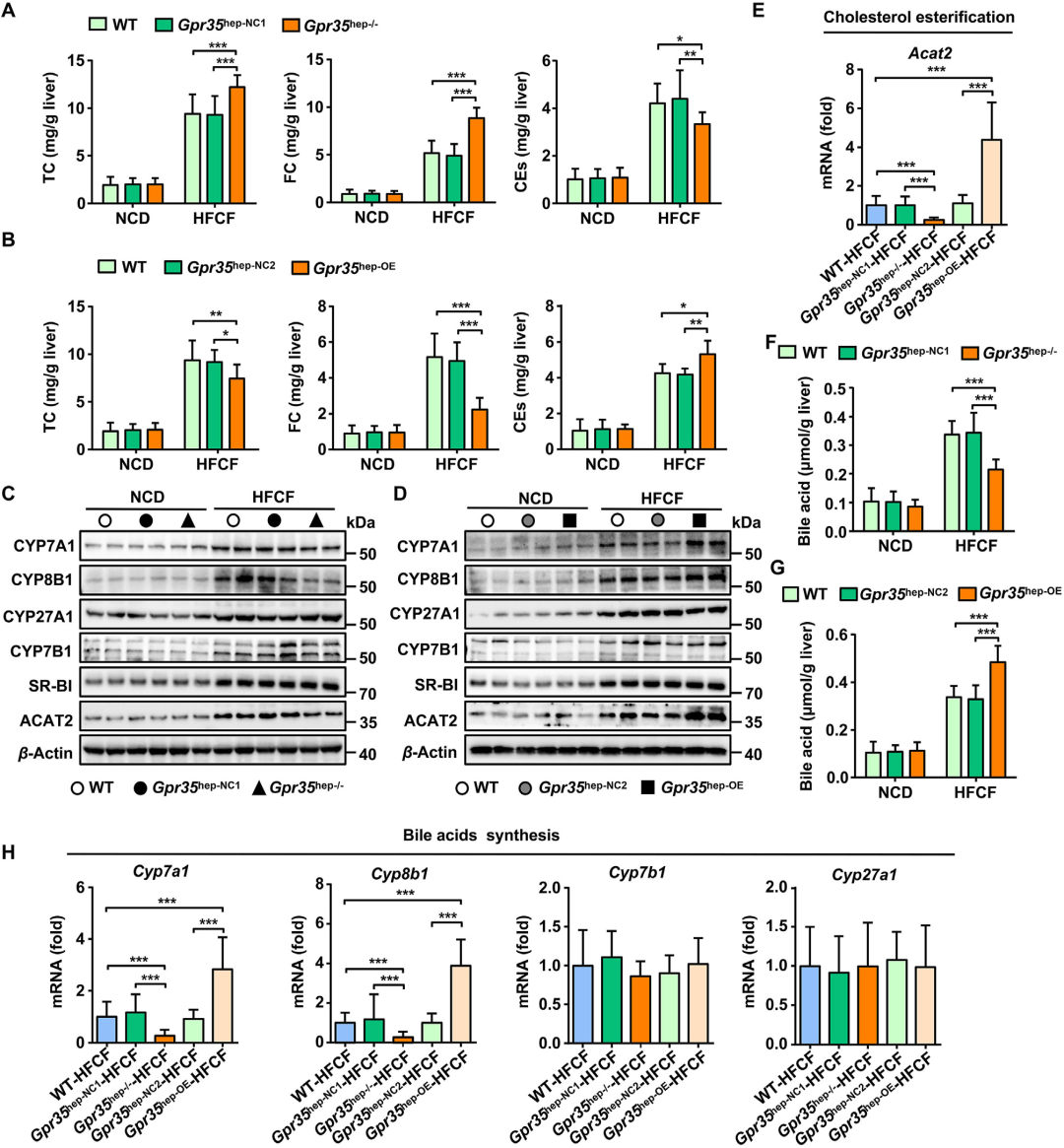
值得注意的是,Gpr35hep-/-小鼠肝脏中的总胆固醇(TC)和FC水平显著升高,而胆固醇酯(CEs)水平降低。相反,Gpr35hep-OE小鼠肝脏中的TC和FC水平下降,CEs水平升高。胆固醇酯化是肝脏排泄FC的重要途径,肝细胞中ACAT2介导的CEs形成可直接促进肝脏胆固醇分泌至血液中。与CEs水平的变化一致,HFCF饮食喂养的小鼠肝脏中GPR35表达上调了Acat2的蛋白和mRNA表达,表明GPR35促进胆固醇酯化。
肝脏是胆固醇生物合成、摄取、排泄以及转化为胆汁酸的主要场所。作者发现,肝细胞中GPR35的表达变化(过表达或缺失)并不调控肝脏胆固醇的排泄(Abcg5、Abcg8、Abca1)、合成(Hmgcr)或摄取(Ldlr、Srb1/Scarb1)相关基因。除肝脏外,肠道在调节胆固醇水平中也发挥重要作用,其通过介导膳食和胆汁胆固醇的肠道吸收,以及直接经肠道排泄胆固醇来维持全身胆固醇稳态,这些过程受肝-肠轴相互作用的调控。为验证肝细胞GPR35是否影响肠道胆固醇吸收和排泄,研究人员检测了肠道中胆固醇吸收(Npc1l1,编码尼曼-皮克C1样蛋白1—胆固醇转运蛋白)和排泄(Abcg5、Abcg8)相关基因的表达,结果显示肝细胞GPR35表达对肠道胆固醇代谢无显著影响。
在人类中,胆固醇向胆汁酸的转化对维持胆固醇稳态和预防FC蓄积诱导的肝损伤至关重要。与FC水平变化相反,相比HFCF饮食喂养的对照小鼠,Gpr35hep-/-小鼠肝脏胆汁酸水平降低,而Gpr35hep-OE小鼠则升高。细胞色素P450(CYP)酶家族通过参与BAS途径在胆固醇清除中发挥关键作用。在肝细胞GPR35缺失的小鼠中,内质网(ER)BAS经典途径的关键酶CYP7A1和CYP8B1表达下调,而线粒体BAS替代途径相关酶CYP27A1和CYP7B1(表达细胞色素P450家族7亚家族b多肽1)的表达未受影响。相反,肝细胞GPR35过表达则上调了CYP7A1和CYP8B1的表达,表明GPR35通过中性途径(由CYP7A1启动)而非酸性途径(由CYP27A1启动)促进胆固醇向胆汁酸转化。此外,研究者检测了肝脏胆汁酸重吸收(Ntcp)、结合(Bal、Bat、Hnf4a1)及外排(Abcb11、Abcb4、Ostb)相关因子的表达,以及肠道胆汁酸重吸收标志物表达(Osta、Ostb、Ibabp、Asbt),发现这些标志物的表达大多不受肝脏GPR35表达的影响。表明肝细胞中GPR35的表达不影响胆汁酸在肝脏中的重吸收、结合和排泄,也不影响在肠道中的重吸收。
重要的是,在全身性GPR35敲除(Gpr35-KO)小鼠中,研究人员观察到一致的结果:GPR35缺失抑制了HFCF饮食喂养小鼠肝脏中ACAT2介导的胆固醇酯化和CYP7A1启动的BAS途径,加剧了肝脏FC蓄积。综上,这些数据表明,肝细胞GPR35可能通过调控胆固醇稳态(酯化和BAS途径)减轻HFCF饮食诱导的NASH。
图5.GPR35调节肝脏BAS和胆固醇酯化
5.GPR35促进肝脏STARD4表达
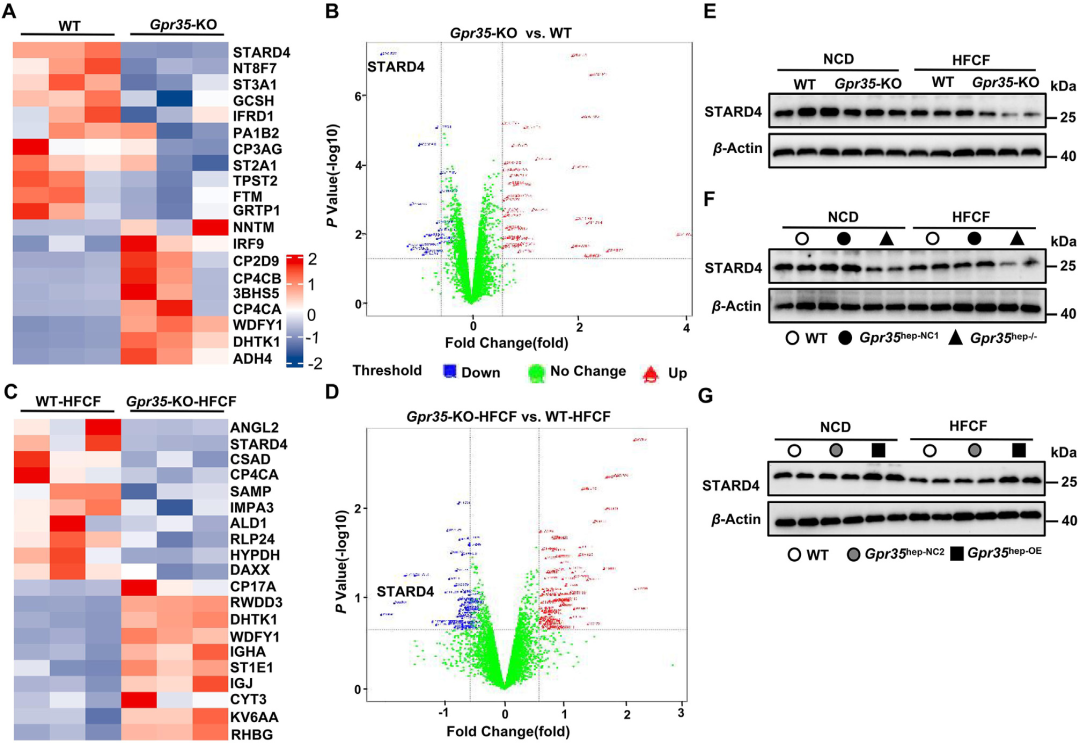
为进一步探索肝细胞GPR35调控胆固醇稳态的机制,研究者通过串联质谱标签(TMT)标记技术对HFCF饮食或NCD喂养的Gpr35-KO与WT小鼠肝组织进行了深度定量蛋白质组学分析,共鉴定出7049种蛋白。通过多重样本检验(假发现率FDR阈值设为0.05)筛选出表达量显著差异的蛋白(变化≥1.5倍且P<0.05)。在NCD喂养的小鼠中,与WT小鼠相比,Gpr35-KO小鼠肝脏有38种蛋白表达上调,18种下调。其中,参与胆固醇胞内转运的蛋白STARD4(UniProt编号:Q99JV5)表现出最显著的表达下调。在HFCF饮食喂养的Gpr35-KO小鼠中,与WT小鼠相比,77种蛋白表达上调,55种下调,且STARD4表达显著降低。通过Western blot进一步验证了Gpr35-KO小鼠肝脏中STARD4的表达低于WT小鼠。STARD4蛋白属于进化保守的类固醇生成急性调节蛋白(StAR)相关脂质转移蛋白家族,广泛表达于多种组织(以肝脏和肾脏水平最高),是一种高效可溶性固醇转运蛋白,对维持胆固醇稳态至关重要。研究表明,STARD4通过将胆固醇运输至ER,促进ACAT2依赖的胆固醇酯化,并提高BAS速率。值得注意的是,STARD4在胆固醇向ER递送中发挥关键作用,而ER富含BAS中性途径的关键酶CYP7A1。实验证实,GPR35促进了肝脏胆固醇向胆汁酸转化及酯化过程。Western blot结果显示,与对照组相比,HFCF饮食喂养的Gpr35hep-/-小鼠肝脏STARD4表达降低,而Gpr35hep-OE小鼠则显著升高,表明GPR35正向调控STARD4表达。因此,作者推测STARD4可能是GPR35通过维持胆固醇稳态在肝细胞中发挥抗脂肪性肝炎保护作用的关键介质。
图6.GPR35促进肝脏STARD4的表达
6.STARD4是GPR35介导的HFCF饮食诱导脂肪性肝炎保护作用的关键调控因子
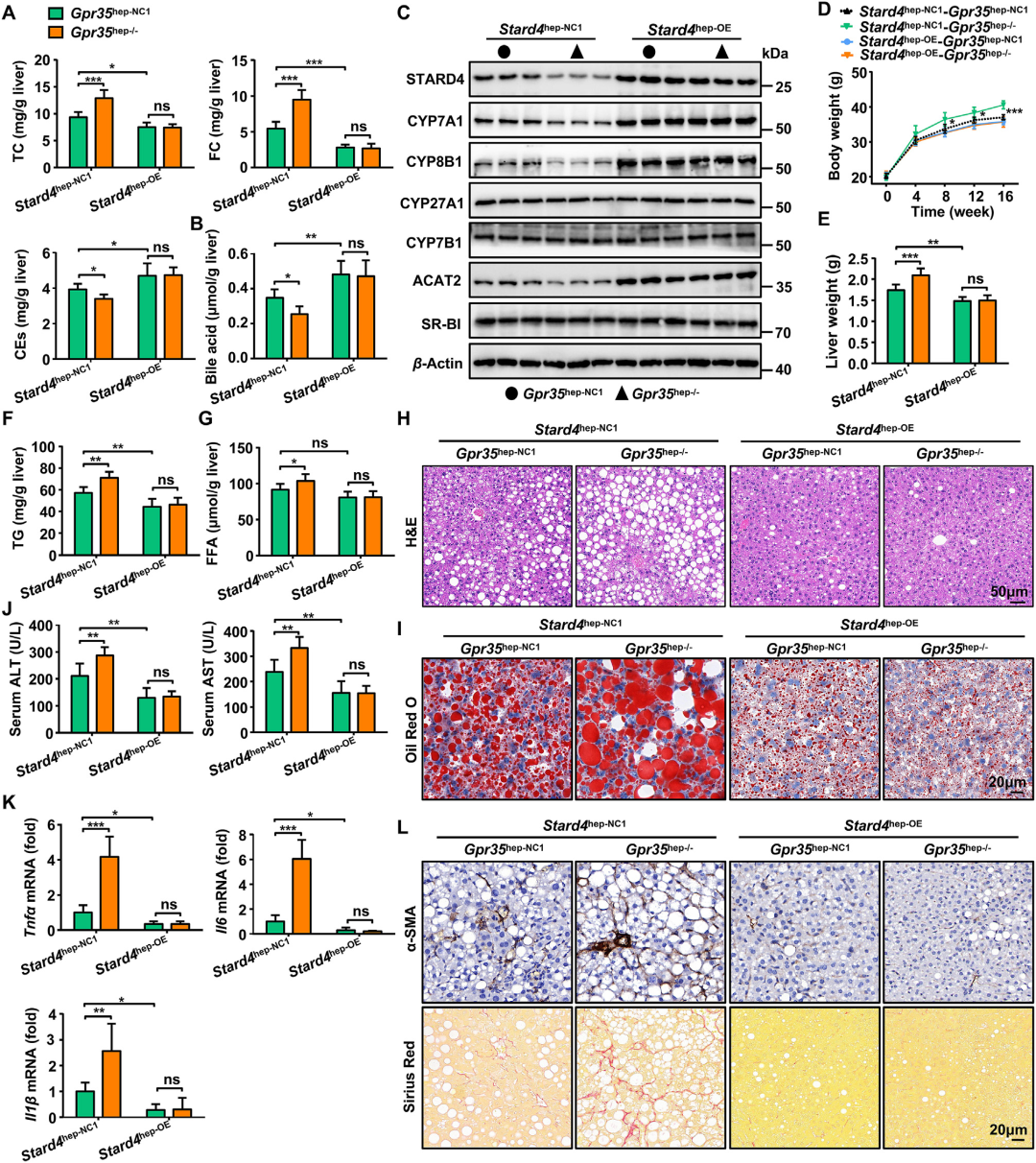
为验证上述假说,研究人员向Gpr35hep-NC1或Gpr35hep-/-小鼠静脉注射AAV8-TBG-m-Stard4-3xFlag-mCherry病毒以增加STARD4表达,随后饲喂HFCF饮食16周。结果显示,在Stard4hep-NC1小鼠中,肝细胞GPR35敲除导致肝脏TC和FC水平升高,CEs及胆汁酸水平降低;而在Stard4hep-OE小鼠中,这些变化被完全逆转。肝细胞GPR35敲除引起的CYP7A1、CYP8B1和ACAT2表达下调在Stard4hep-OE小鼠中得以恢复。与此一致,在Stard4hep-NC1小鼠中,肝细胞GPR35缺失加剧了脂肪性肝炎表型(表现为体重、肝脏重量、肝脏TG和FFA含量增加,肝脏脂肪变性、肝损伤及纤维化加重),而Stard4hep-OE小鼠中未观察到上述恶化效应。
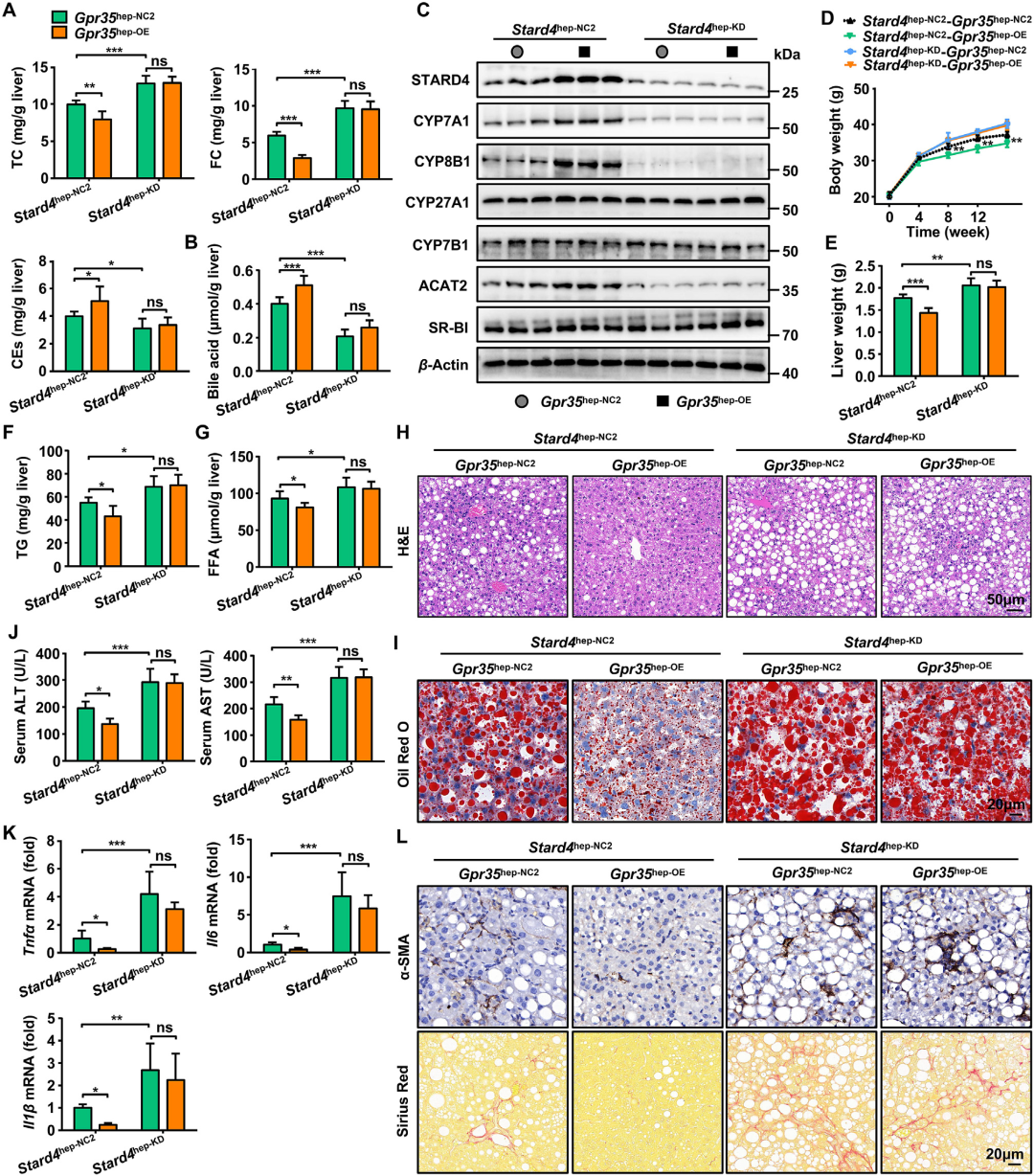
进一步地,研究者向Gpr35hep-NC2或Gpr35hep-OE小鼠静脉注射AAV8-TBG-Mir30-m-shRNA(Stard4)-mCherry病毒以敲低STARD4表达。结果显示,STARD4表达下调完全消除了GPR35过表达对脂肪性肝炎的保护作用,具体表现为胆汁酸生成减少、胆固醇酯化水平下降。然而,无论STARD4过表达或敲低,均未调控肝脏胆固醇排泄(Abcg5、Abcg8、Abca1)、合成(Hmgcr)、摄取(Ldlr、Srb1)或DNL(Acc1、Fasn)相关基因表达。综上,这些数据表明,肝细胞GPR35通过正向调控STARD4表达维持胆固醇稳态,从而发挥对HFCF饮食诱导脂肪性肝炎的保护作用。
图7.STARD4功能的增强逆转了GPR35缺失导致的脂肪性肝炎的加重
图8.STARD4的缺失消除了GPR35对脂肪性肝炎的保护作用
7.肝细胞GPR35通过ERK1/2-CREB信号通路诱导STARD4表达
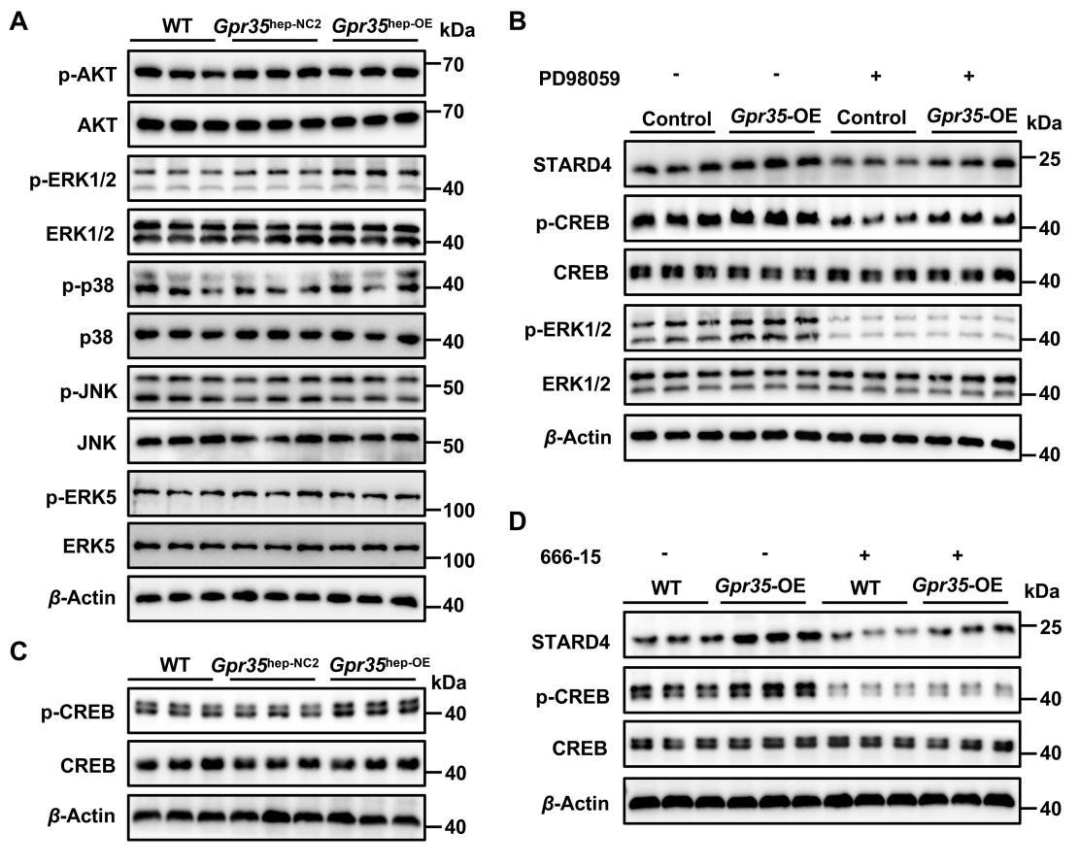
当配体结合后,G蛋白偶联受体(GPCRs)可通过四类G蛋白及其他相互作用蛋白激活多样化的下游信号通路。在GPCR介导的胞内信号传导中,四类丝裂原活化蛋白激酶(MAPK)级联反应是核心组成部分,包括细胞外信号调节激酶(ERK)、c-Jun氨基末端激酶(JNK)、p38 MAPK和大分子MAPK(BMK1/ERK5)。MAPK信号通路的典型特征是激酶在刺激依赖性条件下发生核转位,进而调控转录、细胞代谢及应激相关凋亡的胞内急性反应。本研究发现,与WT及对照组小鼠相比,HFCF饮食喂养的GPR35过表达小鼠肝脏中磷酸化ERK1/2(p-ERK1/2)水平显著升高,而其他MAPK通路成员(p-p38、p-JNK、p-ERK5)及p-AKT水平未发生明显变化。这一结果与既往研究报道的犬尿喹.啉酸(Kyna)/GPR35在肠上皮细胞和脂肪细胞中诱导ERK1/2磷酸化的现象一致。此外,在PA&OA处理的AML-12细胞(小鼠正常肝细胞)中,ERK1/2抑制剂(PD98059)显著逆转了GPR35对STARD4蛋白表达的诱导作用,表明ERK1/2通路参与了GPR35调控STARD4的过程。
在脂肪细胞中,Kyna/GPR35信号通过激活ERK1/2-CREB通路诱导下游基因表达。已有研究证实,ERK1/2和p38介导的CREB信号可上调(StAR)的转录水平,但CREB是否调控STARD4表达尚不明确。为进一步探究GPR35-ERK1/2通路中CREB的作用,本研究通过实验发现:在HFCF饮食小鼠肝脏中,GPR35诱导CREB磷酸化;而在PA&OA处理的AML-12细胞中,ERK1/2抑制剂可抑制这一磷酸化过程。同时,CREB特异性抑制剂(666-15)能够显著阻断GPR35对STARD4蛋白表达的促进作用。综上,在NAFLD模型中,肝细胞GPR35通过激活ERK1/2-CREB信号通路,最终上调STARD4的表达水平。
图9.肝细胞GPR35通过ERK1/2-CREB信号通路诱导STARD4表达
8.GPR35药理学激动剂Kyna通过激活肝脏受体预防HFCF饮食诱导的小鼠脂肪性肝炎
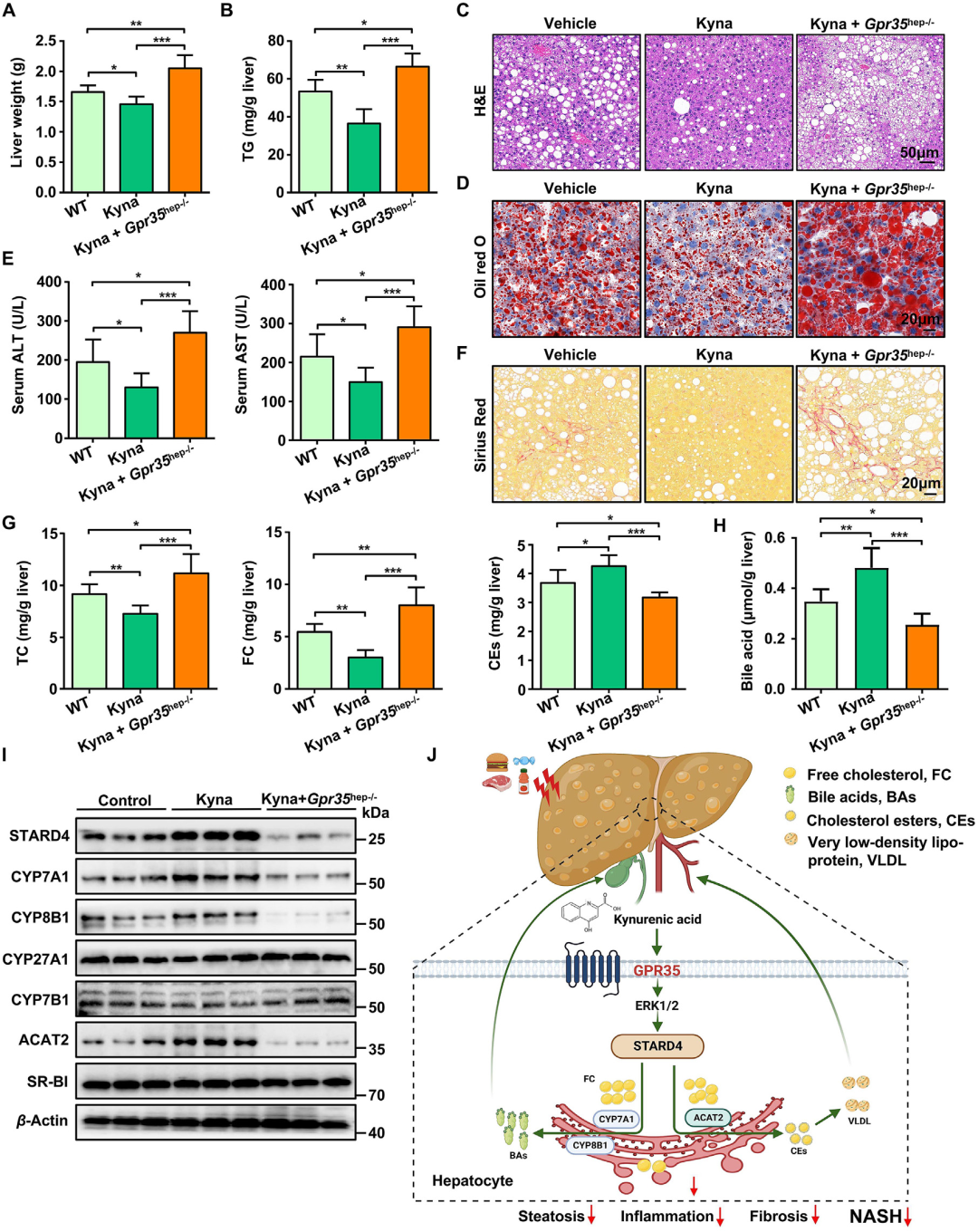
Kyna是孤儿受体GPR35的内源性激动剂。既往研究表明,Kyna可通过激活脂肪组织中的GPR35增加能量消耗。本研究进一步探究Kyna是否能抑制HFCF饮食诱导的肝脏脂肪性肝炎。研究人员将WT小鼠和肝细胞特异性GPR35敲除(Gpr35hep−/−)小鼠饲喂HFCF饮食8周,待其出现显著体重增加和糖耐量受损后,每日腹腔注射Kyna(5 mg/kg体重),并继续HFCF饮食8周。结果显示:与溶剂对照组相比,Kyna处理的WT小鼠肝脏重量和TG含量显著降低。长期Kyna干预改善了HFCF饮食16周诱导的肝脂肪变性、损伤及纤维化。而肝细胞中GPR35表达的缺失阻断了Kyna的保护作用。Kyna干预显著降低了肝脏TC和FC水平,同时增加了CEs和胆汁酸含量。这一效应与Kyna介导的胆固醇代谢相关蛋白(CYP7A1、CYP8B1和ACAT2)表达上调密切相关。当敲除Kyna的受体GPR35后,Kyna对胆汁酸生成和胆固醇酯化的促进作用完全消失。此外,与GPR35过表达小鼠的结果一致,Kyna处理激活了肝脏中的ERK1/2-CREB-STARD4信号通路。综上,Kyna通过激活肝脏中的GPR35受体,抑制HFCF饮食诱导的脂肪性肝炎,其机制涉及胆固醇代谢重编程及下游信号通路的协同调控。
图10.GPR35激动剂Kyna预防HFCF饮食诱导的小鼠脂肪性肝炎
总结
本研究揭示,肝细胞GPR35是NASH中胆固醇稳态的关键调控因子,其功能包括:(1)激活肝脏BAS的经典途径;(2)通过胆固醇酯化促进胆固醇外排。降低FC水平可减轻脂毒性。因此,靶向肝细胞GPR35受体或成为治疗NASH的潜在策略。本研究发现,GPR35的内源性激动剂Kyna能够通过促进胆固醇酯化与胆汁酸生成、降低肝脏FC水平,显著改善实验性脂肪性肝炎。机制上,Kyna处理上调HFCF饮食小鼠肝组织中STARD4表达,进而驱动胆固醇代谢关键酶CYP7A1和ACAT2的表达。值得注意的是,Kyna对肝脏胆固醇稳态及STARD4-CYP7A1/ACAT2轴的调控作用完全依赖于GPR35受体的存在。这一发现为开展靶向GPR35激动剂的临床试验提供了理论依据,有望为NASH治疗开辟新途径。
博士生魏小丽、硕士生殷凡、吴苗苗为本文共同第一作者,王学富教授、王华教授为本文共同通讯作者,安徽医科大学第一附属医院为第一完成单位。本研究受以下基金项目资助:国家自然科学基金杰出青年科学基金项目(82225008)、国家自然科学基金面上项目(82070608)、安徽省自然科学基金青年项目(2108085Y28)及安徽医科大学校科研提升计划项目(2019xkjT007)。
通讯作者介绍:
王学富,博士,教授,药学科学学院(原药学院)。研究方向:(1)肿瘤免疫逃逸机制与免疫治疗;(2)肝脏炎症损伤机制与关键靶标。
王华,主任医师,二级教授,博士生导师,安徽医科大学第一附属医院肿瘤内科副主任,兼肿瘤免疫病区主任;教育部抗炎免疫药物重点实验室主任,炎症免疫性疾病安徽省实验室执行主任;安徽医科大学科技产业部部长。1999年毕业于安徽医科大学医学系临床医学专业,2005年获安徽医科大学临床药理研究所药理学博士学位;2007年至2014年在美国国立卫生研究院(NIH)酒精滥用与依赖研究所(NIAAA)肝病实验室以博士后和访问学者身份开展科学研究。主要研究方向为肝损伤炎症与修复、肿瘤免疫,及相关药物研发。主持科技部国家重点研发、重大专项和国家自然科学基金委青年科学基金项目(A类,B类),联合基金项目等。代表作发表在Hepatology,Journal of Hepatology,Gut等杂志,担任多种杂志的编委包括Hepatology,Gastroenterology等,获安徽省自然科学奖二等奖和安徽省青年科技奖。
本文使用的病毒产品,列表如下:


了解产品及服务
请扫码添加客服微信:BrainVTA2020

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK