2026-06-30 阅读量:14
阿来替尼是第二代ALK酪氨酸激酶抑制剂,为ALK阳性晚期非小细胞肺癌(NSCLC)的一线标准治疗方案,凭借优异的血脑屏障穿透能力与长期生存获益在临床广泛应用。但临床数据显示其不良反应负担显著:34.2%的患者出现肝功能异常,其中3级及以上肝损伤占5.3%;13.8%的患者出现皮疹等皮肤毒性,严重者需减量或停药,直接影响治疗依从性与疗效。
此前研究仅分别探讨肝毒性或皮肤毒性的独立机制,二者并发的内在关联尚不明确。已有研究证实,自噬异常参与多种靶向药物的器官损伤;与此同时,肝脏是血清生物素酶(BTD)的主要合成与来源器官,BTD负责将结合型生物素分解为游离生物素以维持全身生物素循环,肝功能障碍患者血清BTD活性显著下降;而生物素(维生素B7)缺乏的典型表现包含皮肤角化屏障异常。由此推测,肝损伤引发的BTD-生物素轴紊乱,可能是介导肝-皮肤器官间毒性串扰的潜在纽带。然而,该通路是否为阿来替尼多器官毒性的共同核心机制,目前尚未阐明。本研究旨在揭示阿来替尼诱导肝毒性与皮肤毒性的分子机制,探索可临床转化的干预策略。
二 研究概况
浙江大学药学院罗沛华教授、颜皓博士团队于2026年5月在自噬领域权威期刊Autophagy发表题为Liver-originated selective autophagy of BTD by alectinib underlies concurrent hepatotoxicity and dermatotoxicity的研究成果。该研究首次提出「原发肝损伤继发远端皮肤毒性」的器官间串扰新范式,明确肝脏选择性自噬降解BTD是毒性传导的核心机制,并验证了外源性补充生物素的干预价值。

三 核心铺垫:自噬介导阿来替尼诱导的肝细胞损伤
本部分为后续肝脏靶向AAV的机制验证奠定表型基础,通过体内外实验确认肝损伤特征、自噬的驱动作用及肝-皮肤毒性的时序差异。
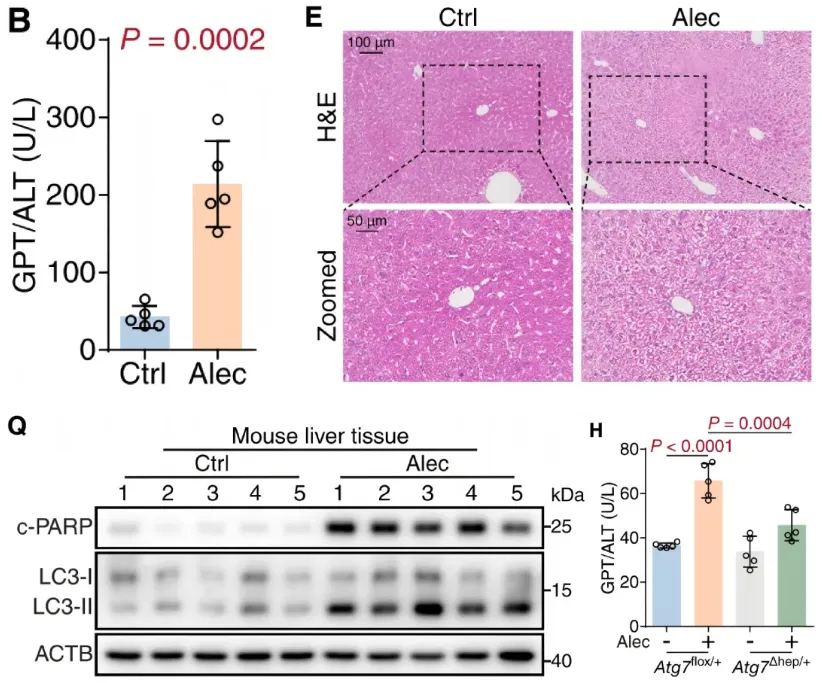
图1 阿来替尼诱导小鼠肝损伤且自噬为上游驱动因素(modified)
阿来替尼给药后小鼠血清ALT升高、肝组织出现水样变性,同时自噬标志物LC3-II与凋亡标志物剪切活化型PARP(c-PARP)同步上调;肝细胞特异性Atg7(自噬体形成的核心必需基因)杂合敲除可显著降低肝酶升高幅度,提示自噬是阿来替尼诱导肝损伤的上游驱动因素,为后续肝脏靶向AAV验证BTD-生物素轴奠定表型基础。
1.体内外肝毒性表型确认
C57BL/6J小鼠每日灌胃500 mg/kg阿来替尼(该剂量为初始表型验证剂量,后续机制及干预实验因动物耐受问题均调整为300 mg/kg),持续4周后血清谷丙转氨酶(GPT/ALT)、谷草转氨酶(GOT1/AST)、总胆红素(TBIL)显著升高;肝组织出现严重水样变性,胶原沉积增加、纤维化程度加重;透射电镜可见肝细胞核固缩,同时胞质内大量自噬空泡累积,提示凋亡与自噬同时发生。
体外实验显示,阿来替尼浓度依赖性降低人肝细胞系THLE-2、HL-7702及小鼠原代肝细胞存活率。补充数据显示,其活性代谢物M4肝脏暴露量远低于母药,因此研究聚焦阿来替尼母药本身。磷酸化蛋白组学分析显示,差异磷酸化蛋白显著富集于自噬、线粒体自噬通路;WB证实自噬标志物LC3-II、凋亡标志物c-PARP均呈浓度与时间依赖性上调。
2.自噬为肝损伤的上游驱动因素
研究利用腺病毒递送的mCherry-GFP-LC3B双荧光系统证实,阿来替尼可激活完整自噬流;时序实验显示LC3-II在给药1.5 h即显著升高,早于c-PARP的上调,且使用自噬晚期抑制剂(氯喹或巴弗洛霉素A1)阻断自噬流,可显著减弱阿来替尼诱导的细胞凋亡。
进一步采用肝细胞特异性Atg7杂合敲除小鼠(Atg7Δhep/+)进行体内基因验证,对照小鼠为Atg7flox/+基因型小鼠。给药4周后,与对照小鼠相比,敲除小鼠的肝酶升高幅度、组织损伤程度、肝细胞凋亡及氧化应激水平均显著减轻,直接证明肝细胞自噬介导了阿来替尼体内肝损伤。
3.肝损伤先于皮肤毒性出现
时间梯度实验显示,给药2周时小鼠已出现早期肝损伤(血清转氨酶升高),但皮肤组织学尚无异常;给药4周时才出现表皮角质层变薄,KRT5、KRT1、KRT10阳性表皮厚度显著降低,提示出现组织学层面的皮肤屏障受损。该时间差与临床皮疹晚发的规律一致,进一步提示皮肤毒性并非药物直接作用,很可能由肝损伤继发介导,支持肝-皮肤器官串扰的研究假设。
四 核心验证:肝脏靶向AAV证实BTD是多器官毒性的共同枢纽
本部分为全文核心,采用肝脏靶向AAV系统直接验证「肝脏BTD是多器官毒性共同源头」的假说,直观体现肝脏靶向AAV在器官串扰研究中的应用价值。
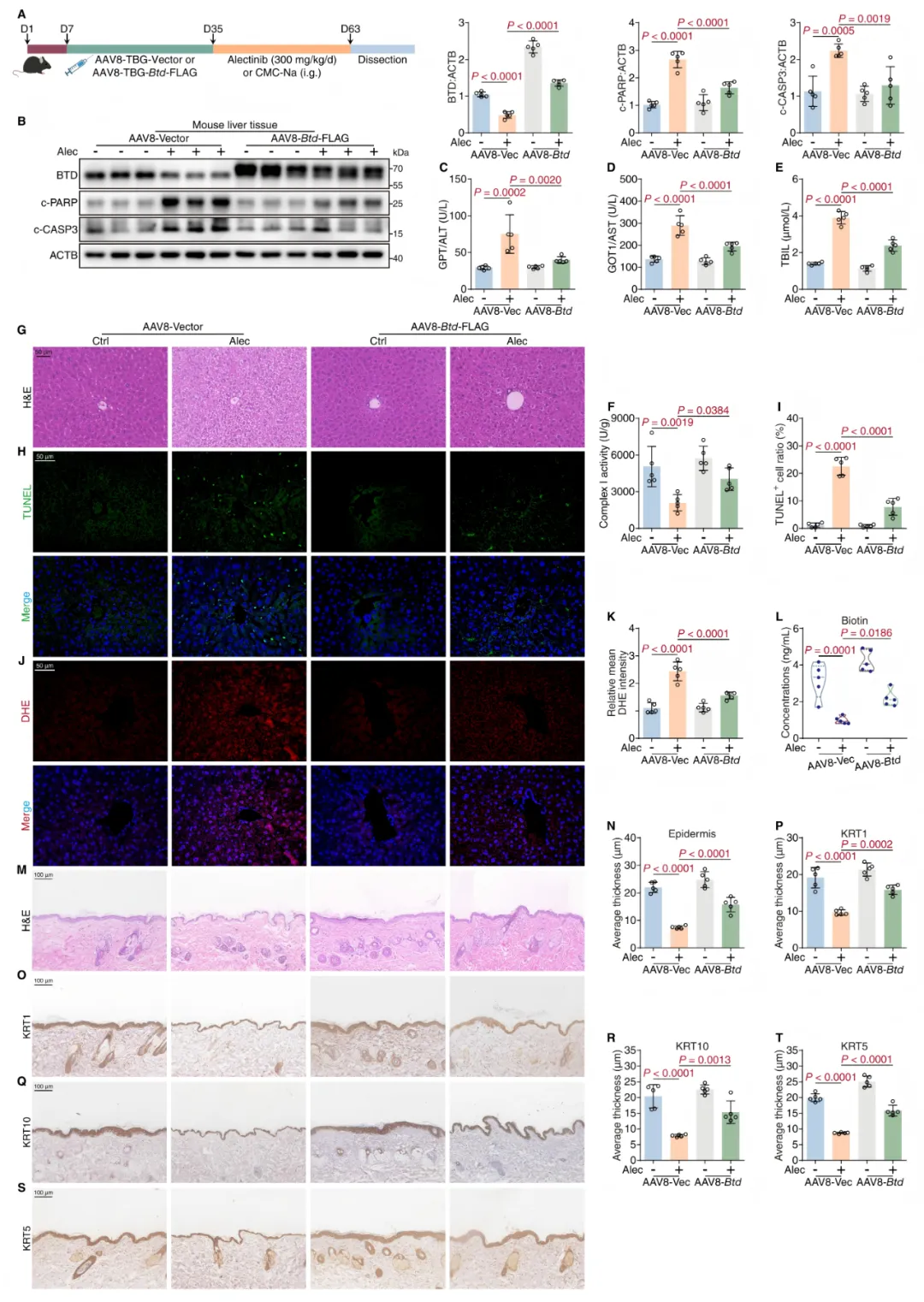
图2 肝脏特异性AAV过表达BTD的实验设计与双器官验证结果
靶向工具设计
研究采用AAV8血清型+TBG肝细胞特异性启动子的经典肝脏靶向体系构建BTD过表达载体(AAV8-TBG-Btd),同时设置空载体(AAV8-TBG-Vector)作为对照组,均通过尾静脉注射给药:
1.AAV8血清型本身具有极强的肝脏嗜性,静脉注射后高度富集于肝组织;
2.TBG(甲状腺素结合球蛋白)启动子具有高度肝细胞特异性,可精准限定目的基因的表达范围;
3.注射后等待4周待基因充分稳定表达,再启动阿来替尼给药实验。
该设计可实现BTD在肝细胞中特异性上调,从而精准验证肝脏来源BTD的全身效应。
双器官同步获益验证
肝脏特异性过表达BTD后,阿来替尼诱导的多器官损伤得到同步逆转:
肝脏层面:血清肝酶、胆红素水平显著回落,肝细胞凋亡与氧化应激减轻,线粒体复合物I活性恢复,肝组织水样变性明显改善;
全身与皮肤层面:血清生物素水平显著回升,表皮变薄与角蛋白(KRT1、KRT10、KRT5)表达下调的表型得到明显修复,皮肤屏障损伤缓解。
该结果直接证明:阿来替尼的皮肤毒性主要继发于肝损伤,而非由药物对皮肤的直接损伤主导;肝脏BTD降解导致的全身生物素缺乏,是连接肝毒性与皮肤毒性的核心纽带,二者通过「肝-皮肤」器官间串扰实现关联。
五 机制延伸:BTD选择性自噬降解的分子调控网络
在明确肝脏BTD是多器官毒性的核心枢纽后,研究进一步通过定量蛋白组学筛选与系列功能验证,完整解析了BTD被选择性自噬降解的分子调控通路:
1.BTD为自噬的特异性毒性底物
对比对照组、阿来替尼组、阿来替尼+巴弗洛霉素A1组的蛋白组,共筛选到45个「药物下调、自噬抑制剂可恢复」的蛋白,结合肝脏高表达与凋亡调控功能,最终锁定BTD为核心候选,KEGG富集也显示生物素代谢为变化最显著的通路。
后续验证排除了转录抑制与蛋白酶体降解途径,明确BTD通过自噬途径降解;BTD耗竭导致细胞内及血清游离生物素下降,进而主要抑制线粒体复合物I活性,降低氧消耗速率与ATP生成,破坏线粒体膜电位并升高ROS,最终触发肝细胞凋亡。
2.NBR1是介导BTD降解的货物受体
在候选自噬受体中,仅敲低NBR1(一种选择性自噬货物受体)可显著逆转阿来替尼诱导的BTD下调与肝细胞凋亡;该表型在3个供体来源的人原代肝细胞中均得到验证,同时敲低NBR1可提升胞内生物素与ATP含量,减少上清肝酶释放。反向验证显示,过表达NBR1可剂量依赖性降低BTD蛋白水平。
机制上,NBR1通过C端UBA泛素结合结构域与BTD相互作用,其中F929、F954位点突变会完全消除二者结合。阿来替尼不增加BTD泛素化水平,而是通过上调NBR1蛋白丰度驱动降解。
3.上游调控:FOXO3激活与NBR1磷酸化协同作用
磷酸化蛋白组学鉴定到NBR1 Ser656位点磷酸化显著增强,该位点多物种高度保守。功能实验证实,Ser656磷酸化可提升NBR1蛋白稳定性,但不影响其与BTD的结合能力。
同时,阿来替尼诱导FOXO3 Ser294磷酸化并入核激活;敲低FOXO3可显著减弱自噬激活,恢复BTD蛋白水平。FOXO3通过转录上调自噬受体SQSTM1激活自噬流,NBR1 Ser656磷酸化则通过增强自身稳定性增加受体丰度,二者协同驱动BTD的选择性自噬降解。
六 转化价值与研究结论
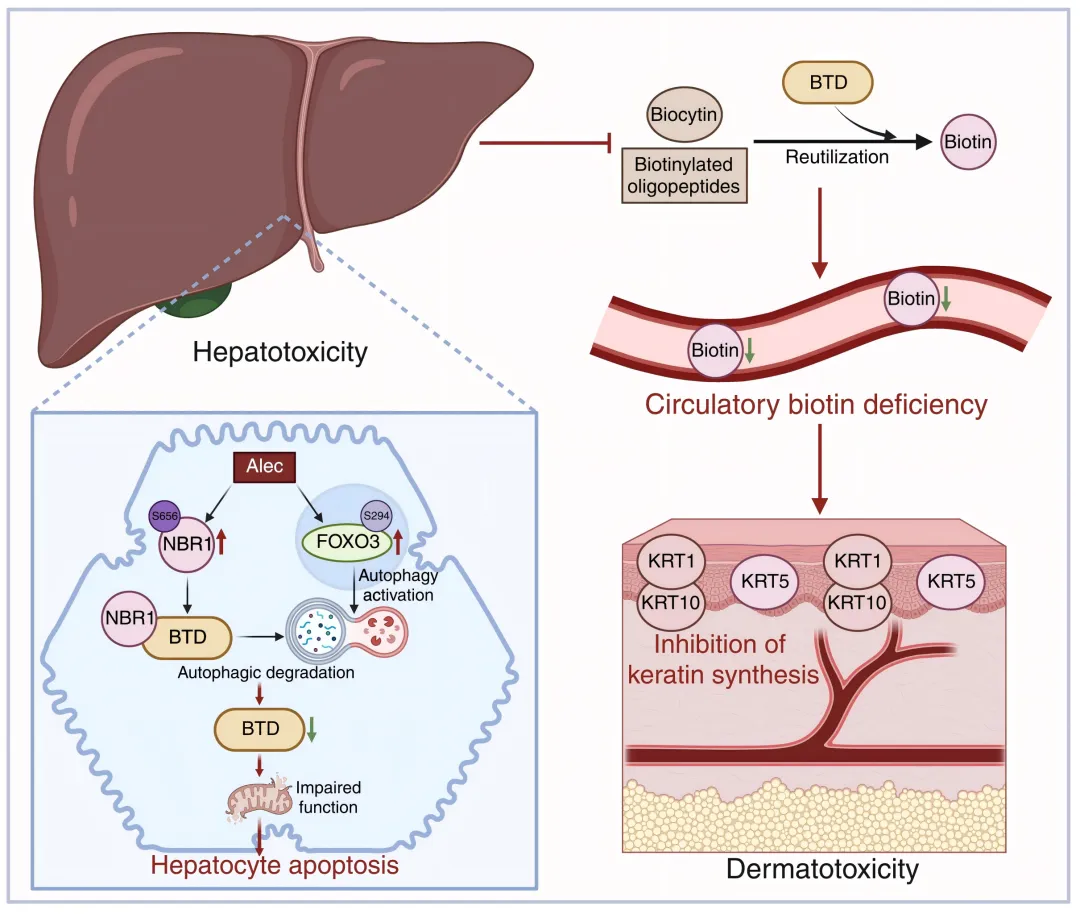
图3 BTD选择性自噬介导阿来替尼肝-皮肤毒性串扰的机制模式图
临床转化潜力
基于机制发现,研究验证了外源性生物素补充的干预价值。体外实验显示,在ALK阳性NSCLC细胞系H3122中,联合1000 nM生物素对阿来替尼的半数抑制浓度(IC50)无显著影响,生物素不削弱阿来替尼的抗肿瘤效果;小鼠体内实验显示,每日补充生物素可同时缓解阿来替尼诱导的肝损伤与皮肤屏障异常,为临床不良反应管理提供了高转化潜力的安全策略。
研究结论
本研究揭示了靶向药物多器官毒性的全新作用范式:阿来替尼并非独立损伤肝脏与皮肤,而是通过诱导肝细胞FOXO3介导的自噬激活,伴随NBR1 Ser656磷酸化稳定性上调,由NBR1作为货物受体介导BTD的选择性自噬降解;肝脏BTD耗竭引发全身生物素缺乏,一方面导致肝细胞线粒体功能障碍与凋亡(原发肝毒性),另一方面引起皮肤角蛋白合成障碍与表皮萎缩(继发皮肤毒性)。
该发现打破了「多器官毒性为药物独立损伤各器官」的传统认知,提出了器官间串扰介导毒性扩散的新机制,同时也解释了临床中皮疹晚于肝损伤出现的现象。其中肝脏靶向AAV工具的精准应用,为器官间互作与多器官毒性研究提供了精准的体内验证策略。
研究局限与展望
本研究仍存在待拓展的方向:NBR1介导BTD降解的结论尚未通过肝细胞特异性基因修饰动物完成体内验证;小鼠皮肤仅呈现组织学改变,未完全重现临床皮疹表型;生物素补充的临床获益仍需真实世界研究进一步确认。此外,研究中发现肠道菌群生物素合成出现代偿性升高,其生理意义尚待进一步探索。
本文使用的病毒产品,列表如下:

上述病毒产品我司均可提供,欢迎咨询!
我司可提供基因过表达、干扰、编辑定制服务,以下为相关产品案例。
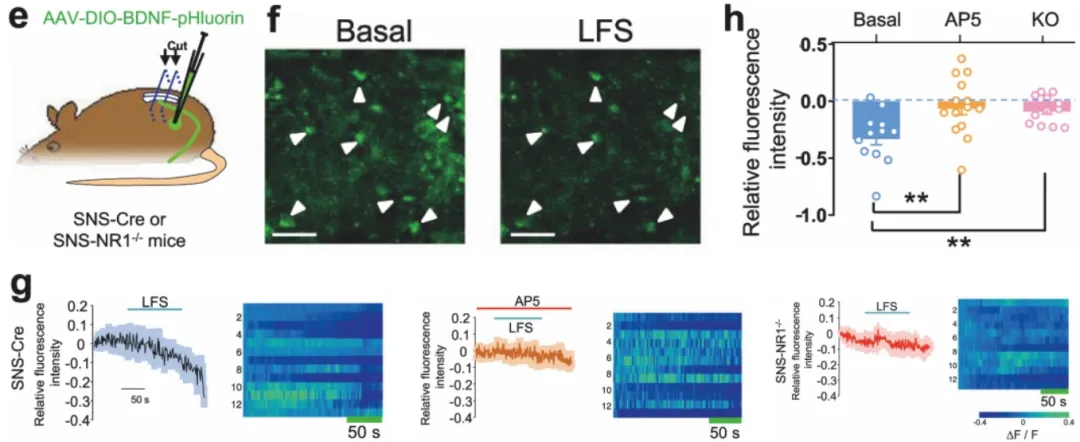
利用AAV介导的BDNF-pHluorin探针表达并结合cre-loxp技术,特异性检测SNS神经元释放的BDNF。(客户文章,Xie RG, Chu WG, Liu DL, et al. Nat Commun. 2022.)。
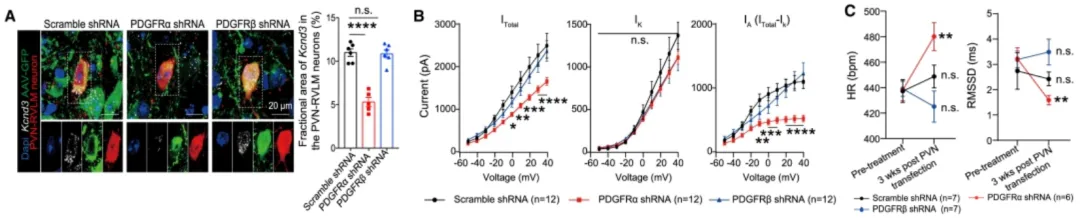
通过AAV介导的shRNA干扰技术对PDGFRa进行敲低,结果显示PVN-RVLM神经元钾电流减少及小鼠心动过速。(客户文章,BiQ, Wang C, Cheng G, et al. Immunity. 2022.)
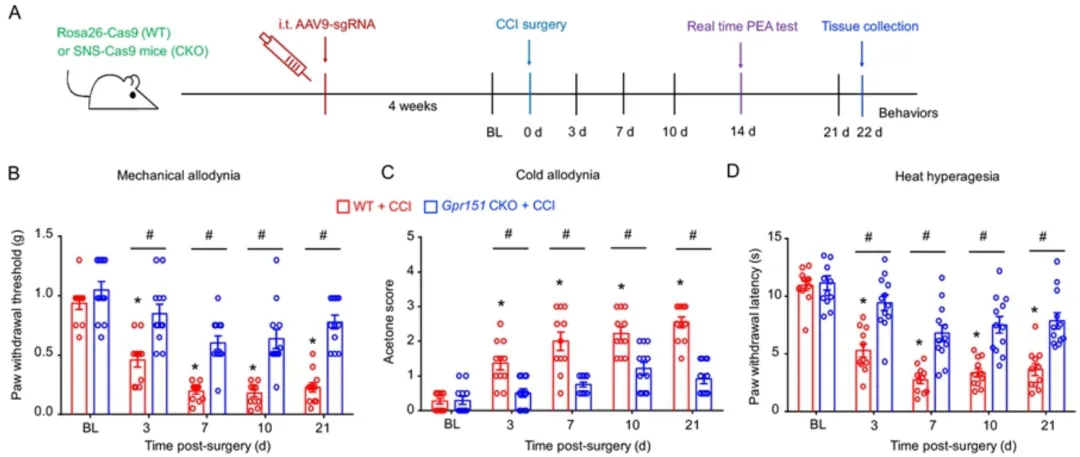
使用CRISPR/Cas9技术在体特异性地敲除DRG中的Gpr151能够有效缓解神经损伤(CCI)引起的神经病理性疼痛。(客户文章,Xia LP, Luo H, Ma Q, et al. Brain.2021.)。
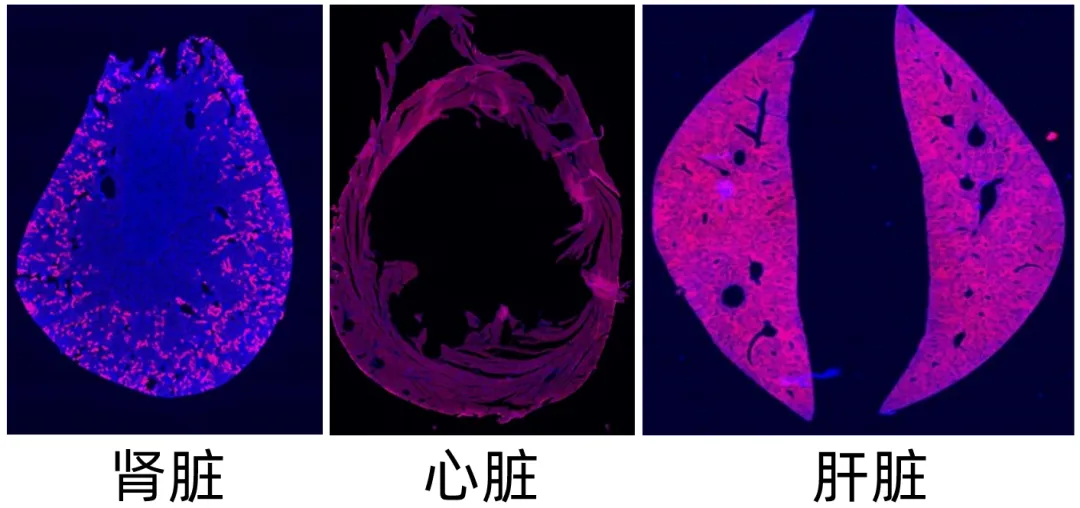
外周脏器病毒感染效果图(rAAV8-mCherry尾静脉注射标记小鼠不同脏器)


市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK
