2026-01-27 阅读量:287
人类大脑需要持续且高效的能量供给,以维持认知功能与日常活动的正常运转。然而,大脑自身的能量储备极少,因此高度依赖膳食来源的营养供给。越来越多的证据表明,饮食在认知功能的塑造过程中发挥着关键作用。其中,富含饱和脂肪的西式高脂饮食(HFD)已成为引发代谢与认知功能双重障碍的重要生活方式诱因。值得注意的是,即便是短期高脂饮食(stHFD)的摄入,也被证实与人类认知表现受损存在关联。此外,流行病学研究显示,代谢综合征患者出现认知功能损伤以及阿尔茨海默病(AD)等神经退行性疾病的风险会显著升高。因此,阐明代谢功能障碍与认知衰退之间的关联机制,对于制定降低此类风险的早期干预策略至关重要。
尽管HFD引发的多种病理特征已被阐明,包括胰岛素抵抗、氧化应激、炎症反应、转录调控异常以及突触可塑性缺陷,但HFD诱导认知损伤的具体细胞与分子机制仍知之甚少。尤其是在HFD摄入的早期阶段,不同脑区及神经元亚型的活动改变规律尚未得到充分探究。明确代谢紊乱过程中最易受损(易感)的神经元群体及其分子层面的改变,是研发针对肥胖、2型糖尿病等代谢性疾病相关认知衰退预防策略的关键环节。
为解答上述科学问题,美国北卡罗来纳大学教堂山分校药理学系宋娟教授团队围绕stHFD诱导的早期神经功能改变开展研究,成果发表于Neuron期刊(IF=15),论文标题为“Targeting glucose-inhibited hippocampal CCK interneurons prevents cognitive impairment in diet-induced obesity”。研究发现,stHFD引发的记忆缺陷由齿状回(DG)胆囊收缩素阳性中间神经元(CCK-INs)过度激活介导。机制层面,该研究首次证实DG CCK-INs为一类新型葡萄糖抑制性神经元;在stHFD条件下,DG区域葡萄糖摄取减少,同时糖酵解关键酶丙酮酸激酶M2(PKM2)磷酸化水平升高,二者共同导致CCK-INs过度激活。恢复DG葡萄糖供给、降低PKM2表达量或抑制其活性,均可使CCK-INs活性恢复正常,并逆转记忆缺陷。基于上述机制,研究进一步验证,在长期饮食诱导肥胖(DIO)小鼠模型中,早期靶向抑制CCK-INs过度激活或PKM2磷酸化,可有效预防认知功能衰退。综上,该研究揭示了饮食代谢应激损伤海马功能的一种此前未被阐明的机制,同时强调DG CCK-INs与PKM2可作为干预代谢紊乱相关认知衰退的潜在治疗靶点。
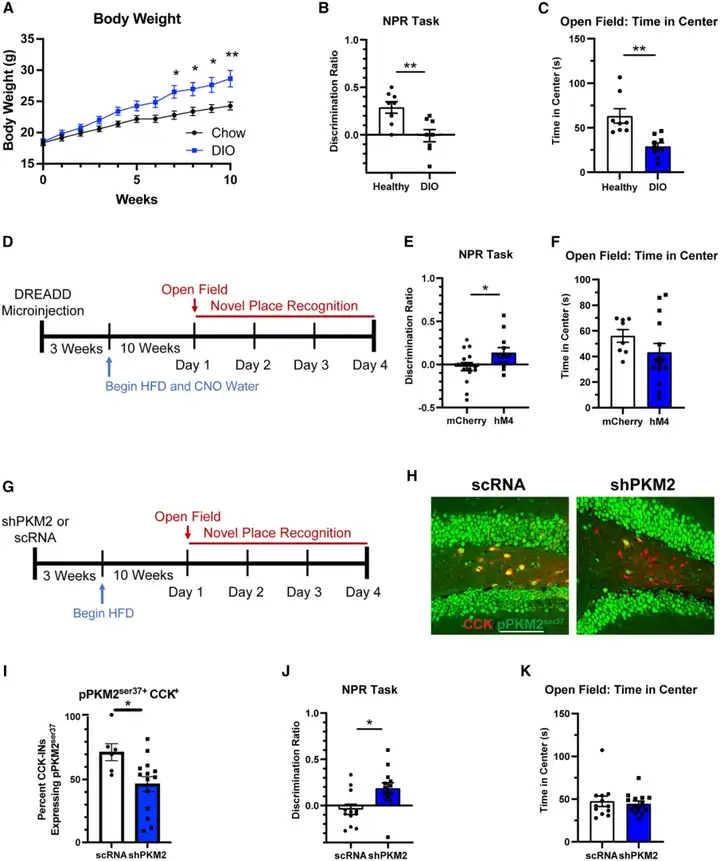
1.海马INs过度激活介导stHFD诱导的认知缺陷
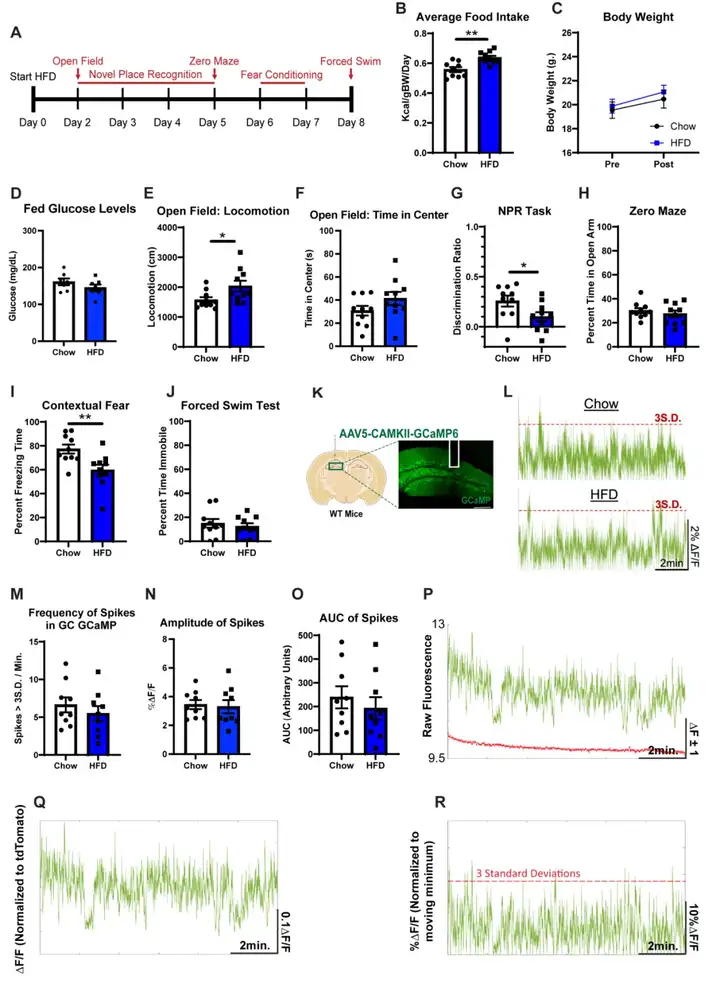
近期研究表明,即使短期摄入HFD也会损害认知功能,而海马对HFD相关损伤尤为易感。为探究上述效应,本研究采用课题组前期建立的系列行为学测试方法,评估小鼠的海马依赖性行为。实验选用8~9周龄野生型(WT)小鼠,给予HFD(脂肪占比58%、碳水化合物25%、蛋白质17%)喂养2天后开展行为学测试。正如预期,HFD组小鼠的热量摄入显著增加;然而,在整个行为学测试阶段,其体重和血糖水平与正常饮食对照组(chow组)相比无显著差异。值得注意的是,HFD组小鼠表现出明显的海马依赖性空间记忆和情境记忆损伤,具体表现为新位置识别实验(NPR)中的辨别率降低,以及情境恐惧条件反射实验(CFC)中的僵直时间缩短。此外,HFD组小鼠在旷场实验(OFT)中表现出运动能力增强。与之相反,各项焦虑样和抑郁样行为学指标未受影响,包括旷场中央区域停留时间、零迷宫(ZM)开放臂停留时间占比以及强迫游泳实验(FST)中的不动时间。上述结果提示,stHFD可选择性破坏海马依赖性认知功能,而对情感行为无显著影响。
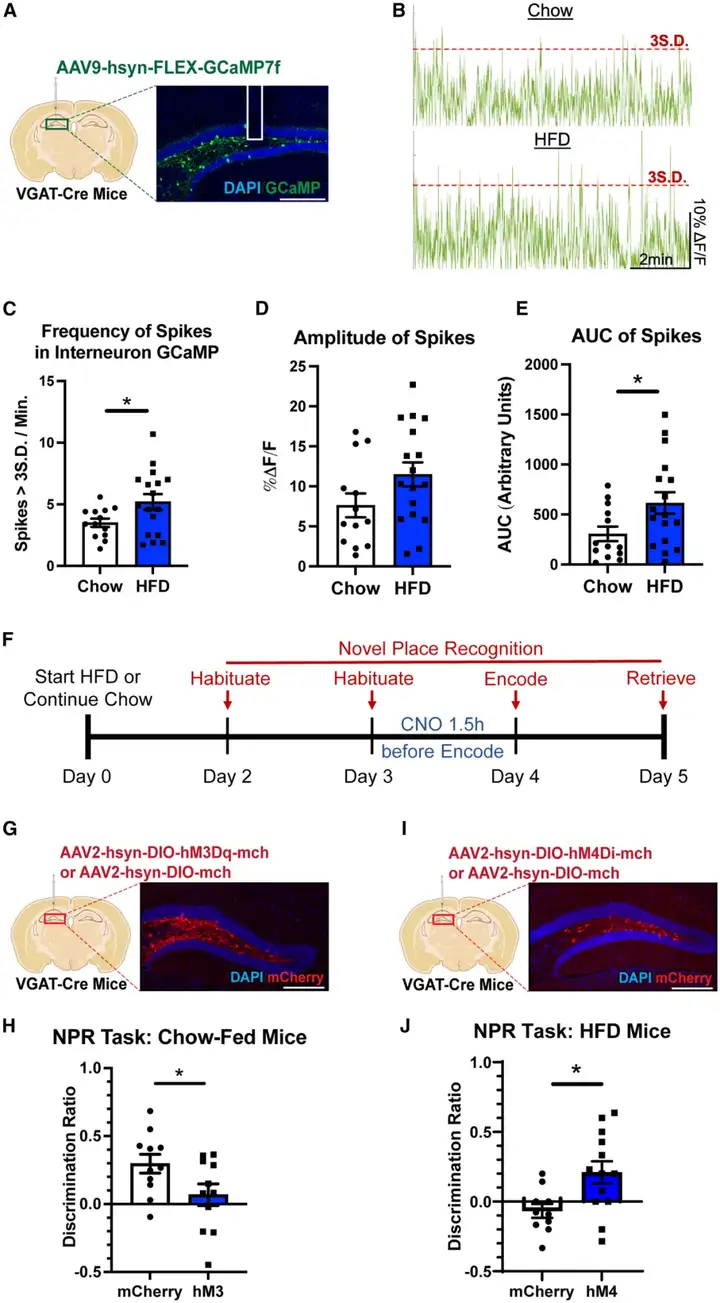
为探究stHFD暴露是否会改变海马环路的动力学特征,本研究聚焦于海马三突触环路的初级输入区——齿状回(DG)。研究人员采用光纤光度记录技术,分别记录chow小鼠与HFD喂养5天的stHFD小鼠DG内两类主要神经元群体——颗粒细胞(GCs)和中间神经元(INs)的神经元群体钙动力学,以此评估该两类神经元的基线活动水平。向WT小鼠或VGAT-Cre小鼠的DG区域注射表达钙指示剂GCaMP的腺相关病毒(AAV),实现对GCs或INs的选择性标记。实验发现,stHFD小鼠的DG INs基线活动显著升高,而GCs活动水平无显著变化。
为明确DG INs过度激活是否参与介导stHFD诱导的认知缺陷,研究人员采用化学遗传学手段激活该类神经元:向VGAT-Cre小鼠体内注射表达兴奋性受体hM3Dq的Cre重组酶依赖性AAV,随后在NPR(一种成熟的DG依赖性记忆检测范式)编码阶段前1.5小时,对小鼠进行氯氮平-N-氧化物(CNO)腹腔注射。结果显示,对chow小鼠的DG INs进行急性激活后,其新位置识别能力显著受损,提示DG INs的过度活动会破坏认知功能。反之,研究人员进一步验证抑制DG INs能否逆转stHFD诱导的认知缺陷:向stHFD喂养的VGAT-Cre小鼠注射表达抑制性受体hM4Di的Cre依赖性AAV,在记忆编码前选择性抑制INs的活动。实验证实,对stHFD小鼠的DG INs进行急性抑制,可显著改善其新位置识别能力。重要的是,所有实验中各组小鼠的摄食量均无差异,排除了代谢因素对实验结果的干扰。综上,本研究证实DG INs过度激活介导了stHFD诱导的认知缺陷,同时揭示了饮食诱导记忆损伤背后的关键环路机制。
图1.DG INs过度激活介导stHFD诱导的认知缺陷
图2.stHFD对行为及GCs基线活性的影响
2.DG胆囊收缩素阳性INs的过度激活介导stHFD诱导的认知缺陷
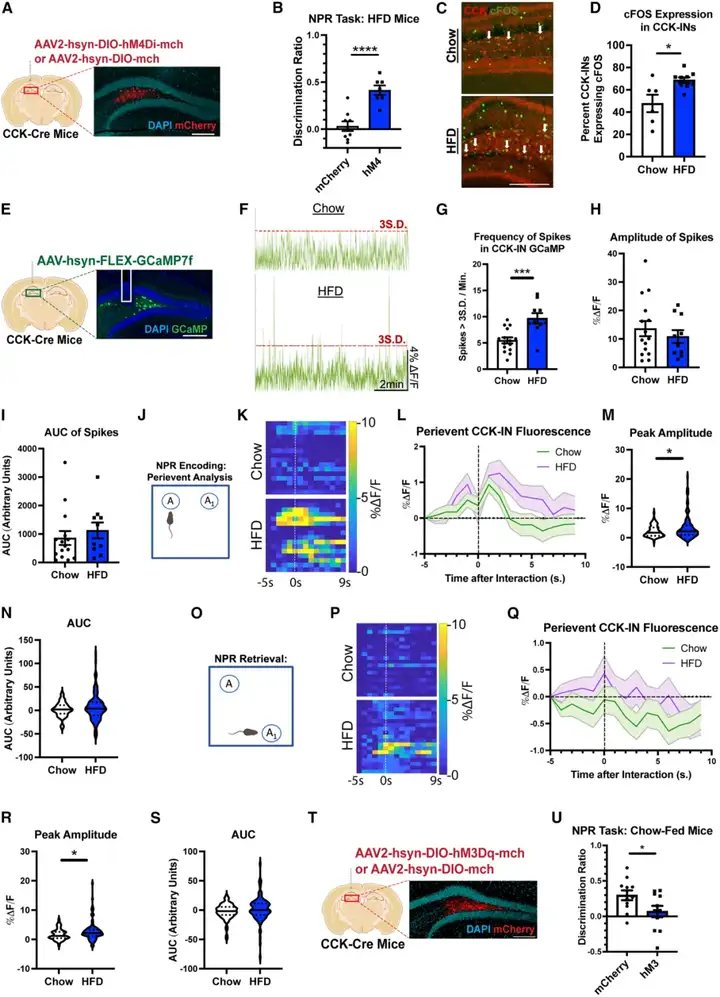
为明确介导stHFD诱导认知缺陷的INs亚型,研究人员向PV-Cre、NPY-Cre及CCK-Cre转基因stHFD小鼠的DG内,注射表达hM4Di的Cre依赖性AAV,从而实现对DG内小白蛋白阳性(PV-)、神经肽Y阳性(NPY-)及胆囊收缩素阳性(CCK-)INs的选择性抑制。结果显示,在记忆编码前1.5小时抑制DG CCK-INs,可显著改善stHFD小鼠在NPR中的记忆表现;而抑制PV-或NPY-INs则无此效应。c-Fos染色与光纤光度记录技术的实验结果为此提供了佐证:stHFD小鼠的DG CCK-INs基线活性显著升高。
为进一步探究认知任务执行过程中CCK-INs的活动特征,本研究在NPR期间同步记录该类神经元的钙动力学。结果显示,在NPR的记忆编码与提取两个阶段,stHFD小鼠在探索物体与空间位置时,其CCK-INs的钙信号反应均显著增强,提示该类神经元的过度激活可能干扰了任务相关的信息加工过程。
为直接验证上述假设,本研究借助hM3Dq在chow小鼠体内实现CCK-INs的急性激活,模拟stHFD小鼠体内该类神经元的过度激活状态。实验结果表明,该操作可显著损害小鼠的记忆表现,进一步证实CCK-INs过度激活在认知功能障碍发生中的作用。需特别指出的是,各组小鼠在实验期间的摄食量无显著差异,这排除了代谢因素对实验结果的干扰。综上,本研究证实:DG CCK-INs的过度激活介导了stHFD诱导的认知缺陷,而抑制该类神经元的异常活性可逆转stHFD相关的记忆损伤。
图3.DG CCK-INs过度激活介导stHFD诱导的认知缺陷
3.CCK-INs活性与葡萄糖可利用度呈负相关
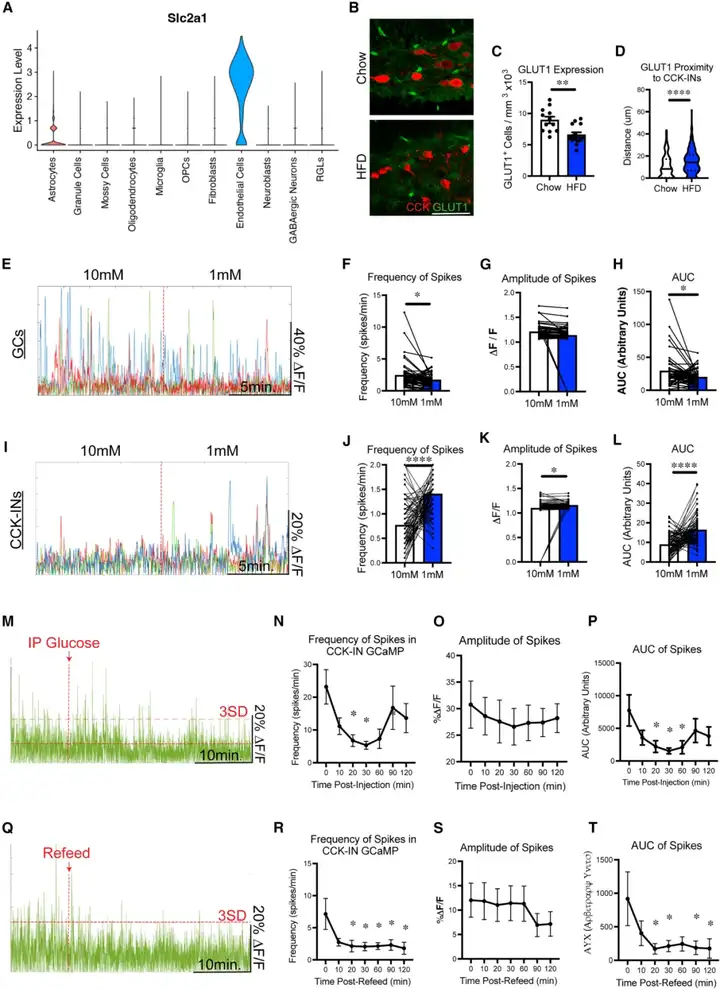
近期有研究指出,葡萄糖转运蛋白1(GLUT1)是一种主要在血脑屏障血管内皮细胞(ECs)中表达的葡萄糖转运载体。小鼠仅接受3天HFD干预后,其全脑范围内(包括海马)的GLUT1表达水平即出现下调。由于GLUT1在介导ECs向神经元转运葡萄糖的过程中发挥关键作用,该表达下调会导致全脑葡萄糖摄取量减少,神经元可利用的葡萄糖水平随之下降。葡萄糖是神经元的主要能量来源,其可利用度降低会对神经元的活性及功能产生显著影响。为进一步探究该效应的脑区特异性,本研究首先通过重新分析已发表的DG组织单核RNA测序(snRNA-seq)数据集,证实GLUT1在DG ECs中存在特异性表达;随后采用免疫组织化学方法,验证stHFD小鼠DG内GLUT1的表达水平显著降低。此外,stHFD小鼠的CCK-INs与最近的GLUT1阳性ECs之间的平均距离显著增加,这提示CCK-INs的葡萄糖转运过程受损。综上,上述研究结果表明,葡萄糖摄取量减少与转运过程受损可能共同导致CCK-INs的葡萄糖可利用度下降。
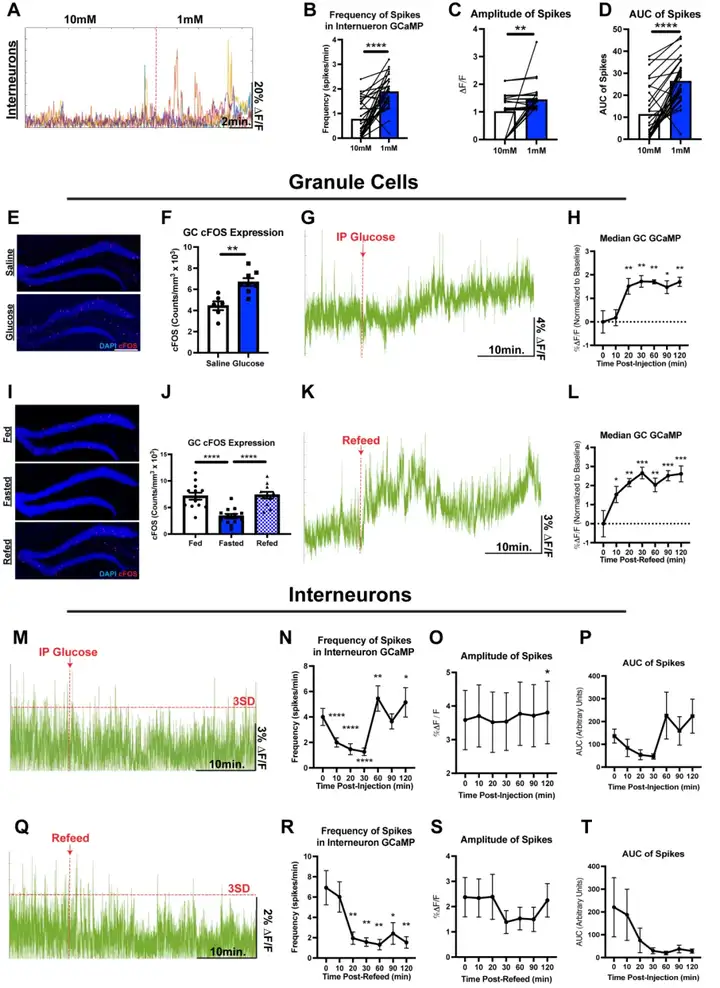
截至目前,葡萄糖可利用度变化对DG不同神经元群体产生的影响尚未得到充分探究。为明确DG神经元(尤其是CCK-INs)是否对葡萄糖敏感,本研究选取在CCK-INs、GCs及γ-氨基丁.酸(GABA)能神经元(VGAT阳性INs)中表达钙指示剂GCaMP的chow小鼠,制备离体急性脑片并开展双光子钙成像实验。实验中,将人工脑脊液的葡萄糖浓度设置为两个浓度梯度:高浓度(10 mM)与低浓度(1 mM),其中低浓度组用于模拟HFD诱导的细胞外葡萄糖可利用度降低状态,高浓度组作为正常对照。有趣的是,研究发现GCs的活性随葡萄糖浓度降低而显著减弱,说明GCs属于葡萄糖兴奋性细胞;与之相反,GABA能神经元与CCK-INs的活性在葡萄糖水平下降后明显增强,这表明DG CCK-INs属于葡萄糖抑制性细胞。基于上述发现,本研究提出如下假设:stHFD小鼠出现的DG CCK-INs过度激活,可能是由脑内葡萄糖摄取受损及GLUT1表达下调引发的葡萄糖可利用度降低所驱动。
为在体验证DG CCK-INs活性与葡萄糖可利用度的负相关关系,本研究对chow小鼠进行过夜禁食以降低其血糖水平,随后检测小鼠在急性葡萄糖注射或复饲2小时前后的GCaMP荧光信号。与离体实验结果一致,葡萄糖注射后20-60分钟内,小鼠CCK-INs的活性即出现显著降低;自由采食复饲同样可在20分钟内抑制CCK-INs的活性,且该抑制效应可持续2小时以上,这可能与记录期间小鼠的多次进食行为有关。值得注意的是,葡萄糖与复饲对GABA能神经元也具有类似的抑制作用,但因VGAT-Cre小鼠品系中被标记的INs存在群体异质性,其响应动力学特征略有差异;而GCs则呈现出相反的变化模式,具体表现为c-Fos表达水平升高及GCaMP基线信号偏移。综上,本研究的离体与在体实验结果共同揭示了DG内存在新型葡萄糖敏感性神经元群体——GCs为葡萄糖兴奋性细胞,而CCK-INs(以及更广泛的DG INs)为葡萄糖抑制性细胞。这些结果为阐明葡萄糖对DG神经环路动力学的调控机制,以及解析改变葡萄糖可利用度的饮食因素对认知功能产生重要影响的作用途径,提供了关键实验依据。
图4.CCK-INs的活性与葡萄糖可利用度呈负相关
图5.GCs与INs的葡萄糖感知功能
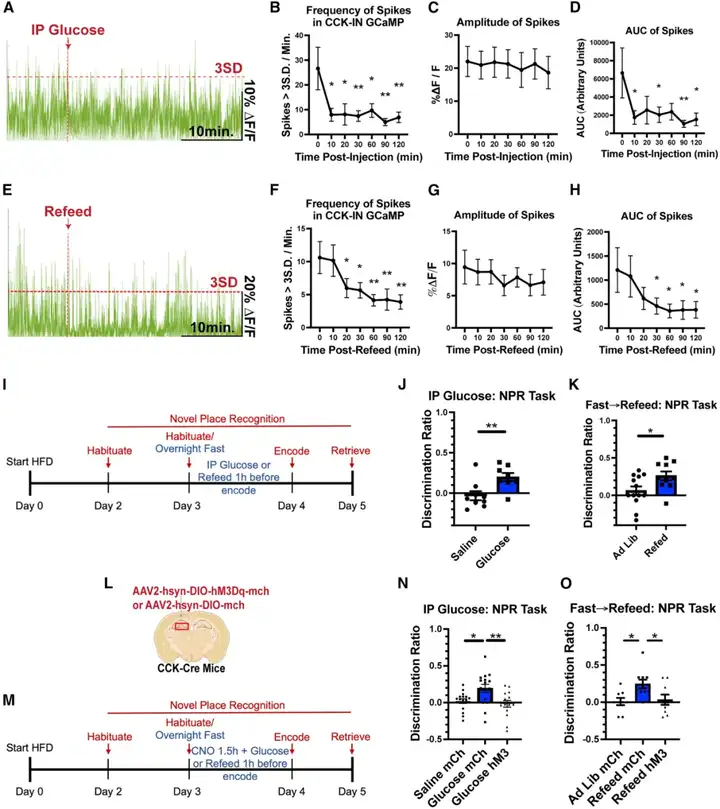
已有研究证实,stHFD小鼠脑内葡萄糖摄取量降低;且既往研究表明,在记忆任务中给予葡萄糖干预对认知功能具有改善作用。基于上述证据,本研究旨在验证以下假设:提高葡萄糖供给可改善stHFD小鼠的认知缺陷。实验方法为:在记忆编码前1小时,对小鼠进行腹腔注射生理盐水或高剂量葡萄糖(2 g/kg body weight),随后通过NPR评估其认知表现。此外,为探索提高葡萄糖供给的替代方案,本研究还设置了自由进食组与禁食复饲组进行对照:将自由进食的stHFD小鼠,与禁食过夜后在记忆编码前1小时复饲HFD的小鼠进行对比,明确急性进食对其记忆表现的影响。结果显示,与chow小鼠类似,腹腔注射葡萄糖与复饲两种干预方式,均能抑制stHFD小鼠内CCK-INs的活性;且这一现象与小鼠记忆表现的改善呈现显著的同步性。
为进一步证实提高葡萄糖可利用度是通过抑制CCK-INs,从而改善stHFD小鼠的记忆缺陷,本研究在记忆编码前、葡萄糖注射或复饲干预前,利用hM3Dq对CCK-INs进行急性激活。结果发现,CCK-INs的激活完全抵消了葡萄糖注射与复饲两种干预方式对记忆的改善效应。这一结果表明,抑制CCK-INs是提高葡萄糖供给,从而恢复认知功能的关键机制。综上,本研究结果为以下结论提供了直接实验证据:通过急性干预提高葡萄糖可利用度,能够缓解stHFD小鼠的CCK-INs过度激活,并改善其记忆表现。
图6.提高葡萄糖可利用度可改善stHFD小鼠的CCK-INs过度激活与认知缺陷
5.PKM2ser37磷酸化水平升高,介导stHFD小鼠CCK-INs过度激活与认知缺陷的关联
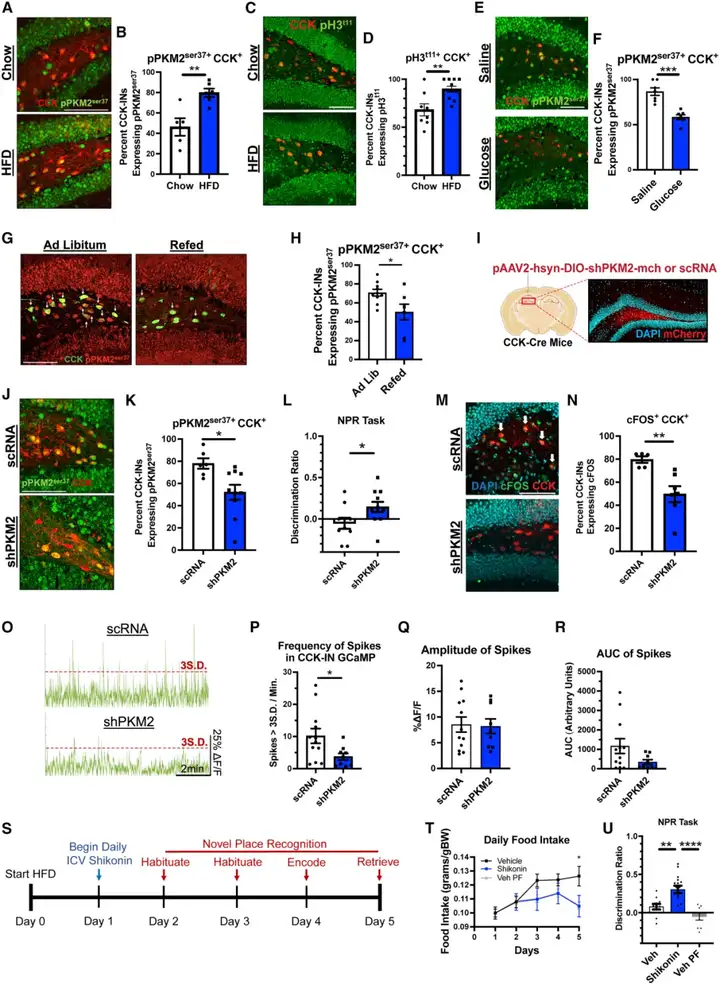
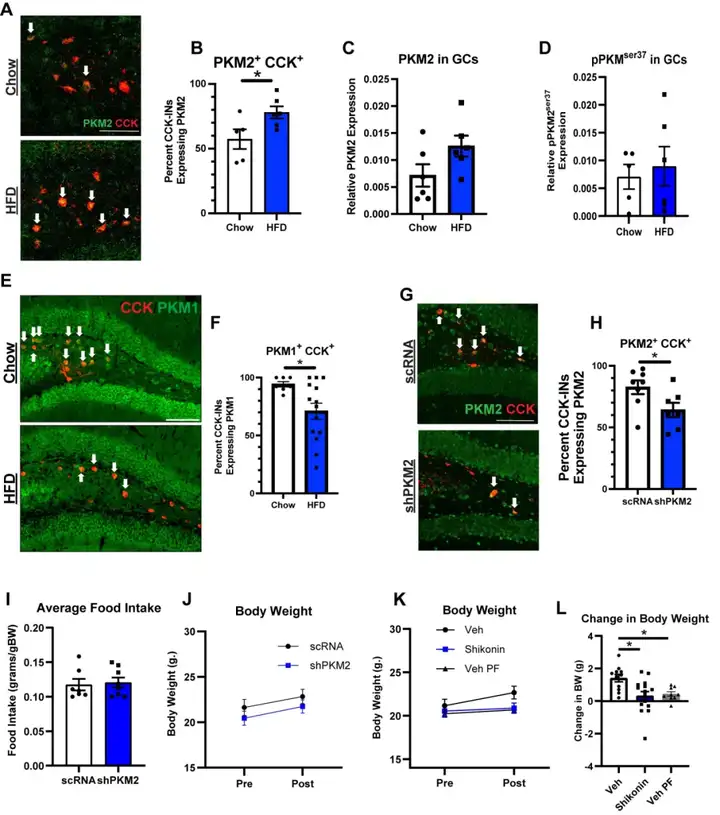
糖酵解酶丙酮酸激酶M2(PKM2)的表达水平常因葡萄糖可利用度降低而上调。当该蛋白的丝氨酸37位点发生磷酸化修饰时,其会发生核转位并驱动转录组改变,进而促进有氧糖酵解过程——这一现象被称为瓦博格效应(Warburg effect),最初在肿瘤细胞中被发现。与此一致,本研究观察到,DG CCK-INs内的总PKM2蛋白表达量显著升高,且更重要的是,丝氨酸37位点磷酸化的PKM2(pPKM2ser37)水平显著上调;而GCs中上述指标无明显变化,提示stHFD可选择性增强DG CCK-INs内PKM2活性。此外,stHFD小鼠的CCK-INs还表现出组蛋白H3苏氨酸11位点磷酸化修饰(pH3t11)水平升高——该位点是核定位PKM2的直接下游靶点;同时伴随丙酮酸激酶M1(PKM1)表达下调,而PKM1正是促进氧化磷酸化过程的同工酶亚型。这些结果提示,stHFD可诱导细胞内信号通路从维持稳态的PKM1活性模式,向核PKM2信号通路模式转变。有趣的是,stHFD小鼠经腹腔注射葡萄糖,或禁食过夜后复饲1小时,其体内pPKM2ser37的表达水平均显著降低,这表明提高葡萄糖供给可作为使pPKM2ser37水平恢复正常的有效手段。
为明确PKM2在stHFD诱导的认知缺陷中发挥的因果作用,本研究采用Cre依赖性AAV,将经过验证的靶向PKM2的短发夹RNA(shPKM2)递送至CCK-INs,实现该细胞群中PKM2的特异性敲低。与乱序(scrambled)shRNA(scRNA)对照组相比,shPKM2处理组小鼠的CCK-INs中,PKM2及pPKM2ser37阳性细胞占比显著降低,证实了敲低效率。值得注意的是,CCK-INs特异性敲低PKM2的stHFD小鼠,在NPR中其记忆表现较对照组显著改善;光纤光度记录技术及c-Fos蛋白表达检测结果显示,该记忆改善效应伴随CCK-INs活性的降低。此外,各组小鼠的摄食量与体重均无显著差异。
为进一步验证抑制PKM2对记忆的挽救效应,本研究采用已在基础研究领域被广泛认可的特异性PKM2抑制剂——紫草素,通过药理学手段靶向抑制PKM2的核转位过程。然而,对stHFD小鼠实施每日侧脑室注射紫草素处理后,小鼠出现摄食量减少及体重下降的意外现象,因此需采用配对喂养模型以排除相关干扰。配对喂养组小鼠的每日供食量与紫草素处理组小鼠的每日供食量完全一致,以此平衡两组间的热量摄入差异。结果显示,尽管已排除摄食量减少带来的干扰,紫草素处理的stHFD小鼠,在NPR中的记忆表现仍显著优于溶媒处理的自由进食组stHFD小鼠及配对喂养组stHFD小鼠。综上,本研究结果表明:stHFD可上调DG CCK-INs中PKM2的表达水平并增强其核内活性,进而导致CCK-INs过度激活与小鼠认知功能缺陷。
图7.PKM2活性增强介导stHFD小鼠CCK-INs过度激活与认知缺陷的关联
图8.CCK-INs中PKM2介导stHFD小鼠的认知功能损伤
6.靶向CCK-INs功能异常的早期干预策略,可预防饮食诱导肥胖小鼠的认知功能衰退
基于在stHFD小鼠模型中取得的创新性发现,本研究进一步采用成熟的饮食诱导肥胖(DIO)2型糖尿病小鼠模型,探究靶向CCK-INs功能异常能否在慢性代谢紊乱模型中预防认知功能缺陷。DIO小鼠经HFD喂养10周,于第7周表现为体重超标。一系列行为学实验检测结果显示,这类小鼠在NPR中的辨别指数降低,且在OFT中央区域的停留时间缩短,提示其空间记忆受损,并表现出轻度焦虑表型。值得注意的是,小鼠的自主活动能力,以及在零迷宫、CFC、FST中的表现均未发生显著变化。尤为关键的是,通过饮水给予CNO,利用hM4Di慢性抑制DG CCK-INs,或在DG CCK-INs中长时程敲低PKM2,均可改善DIO小鼠在NPR中的表现,挽救其空间记忆缺陷。但上述两种干预手段均未改变小鼠在OFT中央区域的停留时间,这表明DG CCK-INs与PKM2对认知功能具有特异性调控作用,而不参与焦虑相关行为的调节。综上,本研究结果表明,靶向代谢损伤诱导的神经元异常实施早期干预,有望缓解代谢性疾病引发的远期认知功能衰退。
图9.靶向HFD诱导早期相关神经元异常的干预措施可预防DIO小鼠的认知衰退
总结
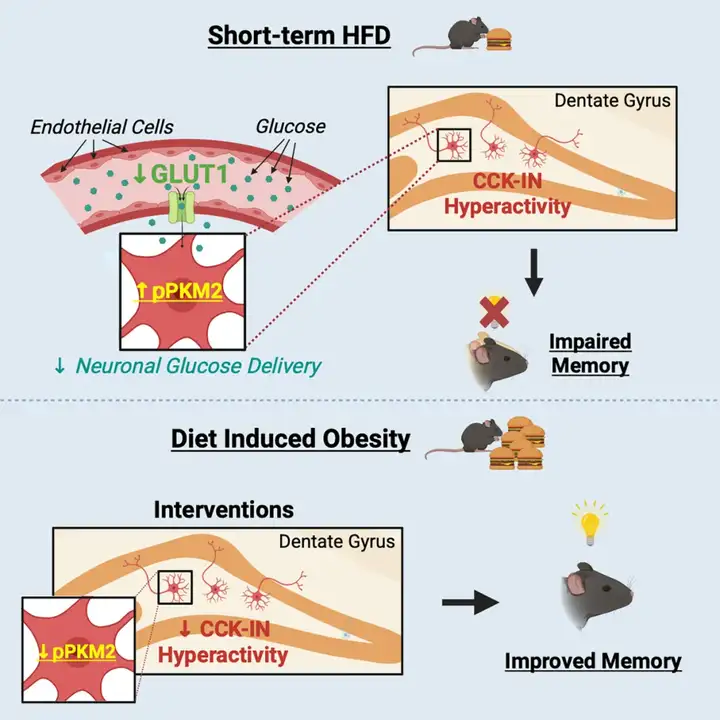
代谢性疾病(尤其是肥胖和2型糖尿病)的发病率日益攀升,其对认知功能的影响已引发广泛关注。尽管已有大量研究证实,HFD摄入与认知功能衰退存在密切关联,但造成这些损伤的具体细胞与分子机制,目前尚未被充分阐明。本研究揭示了一种全新机制——HFD诱导的代谢应激,可通过葡萄糖抑制性DG CCK-INs的过度激活及PKM2介导的代谢重编程,最终损害认知功能。本研究证实,早期代谢干预可有效预防认知功能衰退,这一发现为应对肥胖相关认知衰退带来的日益沉重的疾病负担,开辟了全新的治疗方向。
模式图
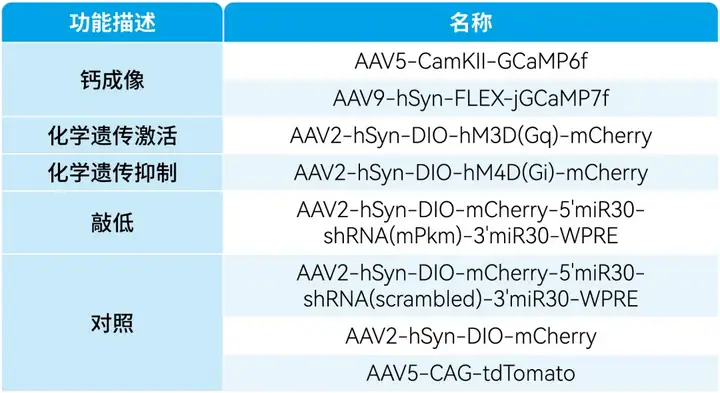
本文使用的病毒产品,列表如下:
扫码添加客服
更多产品及详情欢迎咨询!


市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK