2025-10-16 阅读量:701
心肌肥厚是心脏对多种应激状态的一种反应,如压力超负荷及心肌梗死等。人类患者与动物模型的研究均表明,在大多数情况下,病理性心肌肥厚最终会进展为心力衰竭(HF)。尽管近年来治疗手段取得了进展,但HF仍是全球范围内发病率高、治疗成本高且发病率、致死率持续上升的疾病。为改善临床预后并规避血流动力学风险,亟需研发针对疾病内在机制的新型疗法。
泛素化修饰是一种具有动态性与多面性的翻译后修饰,参与心肌肥厚、缺血性心脏病及HF等常见心脏疾病的病理过程。泛素(Ub)需在泛素化酶的作用下结合到靶蛋白上,这类酶包括Ub激活酶(E1)、Ub结合酶(E2)及Ub连接酶(E3)。在特定E3的催化下,Ub分子可通过其8个位点(M1、K6、K11、K27、K29、K33、K48、K63)中的任意一个进一步发生泛素化,从而形成靶蛋白的多聚Ub链。目前针对这些Ub链类型的大量研究已证实:在病理性心肌肥厚中,K48连接泛素化主要参与蛋白酶体介导的蛋白降解过程,而K63连接泛素化则在细胞信号传导中发挥关键作用。然而,其他类型的多聚泛素化(如K27连接泛素化)在病理性心肌肥厚中的作用仍有待进一步阐明。
含有WW结构域的E3 Ub蛋白连接酶1(WWP1)属于C2-WW-HECT型E3,其结构包含N端C2结构域、4个串联的WW结构域,以及C端负责泛素转移的催化性HECT结构域。WWP1可作为E3,通过作用于含PY基序的蛋白及不含PY基序的蛋白,调控蛋白质转运与降解、转录及信号传导等多种细胞生物学过程。如,心肌细胞特异性过表达WWP1可诱发心律失常与心肌肥厚,提示WWP1功能获得性改变对心脏具有损伤作用。但截至目前,尚未通过功能缺失模型充分揭示WWP1在心肌肥厚中的具体作用,且WWP1介导的K27连接泛素化在心肌肥厚中的作用也亟待探究。
散乱蛋白家族(DVL:DVL1、DVL2、DVL3)在进化上高度保守,是经典及非经典Wnt信号通路中的重要胞质介导分子,在心脏重构过程中发挥关键作用。已有研究证实DVL在心肌肥厚中具有重要功能:DVL可激活钙/钙调蛋白依赖性激酶II(CaMKII),后者进一步磷酸化组蛋白去乙酰化酶4(HDAC4),促使HDAC4核输出;而HDAC4核输出可激活肌细胞特异性增强因子2C(MEF2C),进而启动心肌基因转录,最终导致心肌肥厚。此外,在肥厚心脏中DVL的表达水平升高;散乱蛋白1(DVL1)缺失可延缓压力超负荷诱导的心肌肥厚的发生。在293T细胞(人胚肾293T细胞)中,ITCH与NEDD4L(同属E3)均可通过催化Ub分子以K48连接的方式修饰散乱蛋白2(DVL2),将其靶向至蛋白酶体进行降解,从而调控DVL2的蛋白水平。但目前,心肌肥厚状态下DVL蛋白水平的调控机制仍不明确。
近期,科学家在Circulation期刊(IF=23.603)以“Targeting E3 Ubiquitin Ligase WWP1 Prevents Cardiac Hypertrophy Through Destabilizing DVL2 via Inhibition of K27-Linked Ubiquitination”为题在线发表了研究论文。该研究中,作者发现HF患者及主动脉横向缩窄(TAC)术后小鼠心脏中,WWP1表达水平显著升高。WWP1基因敲除(KO)对压力超负荷诱导的心肌肥厚具有保护作用。机制上,DVL2是WWP1的相互作用蛋白;WWP1通过介导DVL2发生非典型K27连接泛素化修饰维持其蛋白稳定性,从而经DVL2/CaMKII/HDAC4/MEF2C通路加剧心肌肥厚。以WWP1为治疗靶点,通过携带靶向WWP1的短发夹RNA(shRNA),且受心肌肌钙蛋白T(cTnT)启动子调控的腺相关病毒9型(AAV9),在心肌细胞中特异性敲低其表达,可几乎完全消除TAC诱导的心肌肥厚及心功能障碍等负面影响。综上,WWP1是压力超负荷诱导心脏重构的潜在治疗靶点。

1.WWP1在心肌肥厚过程中的表达特征
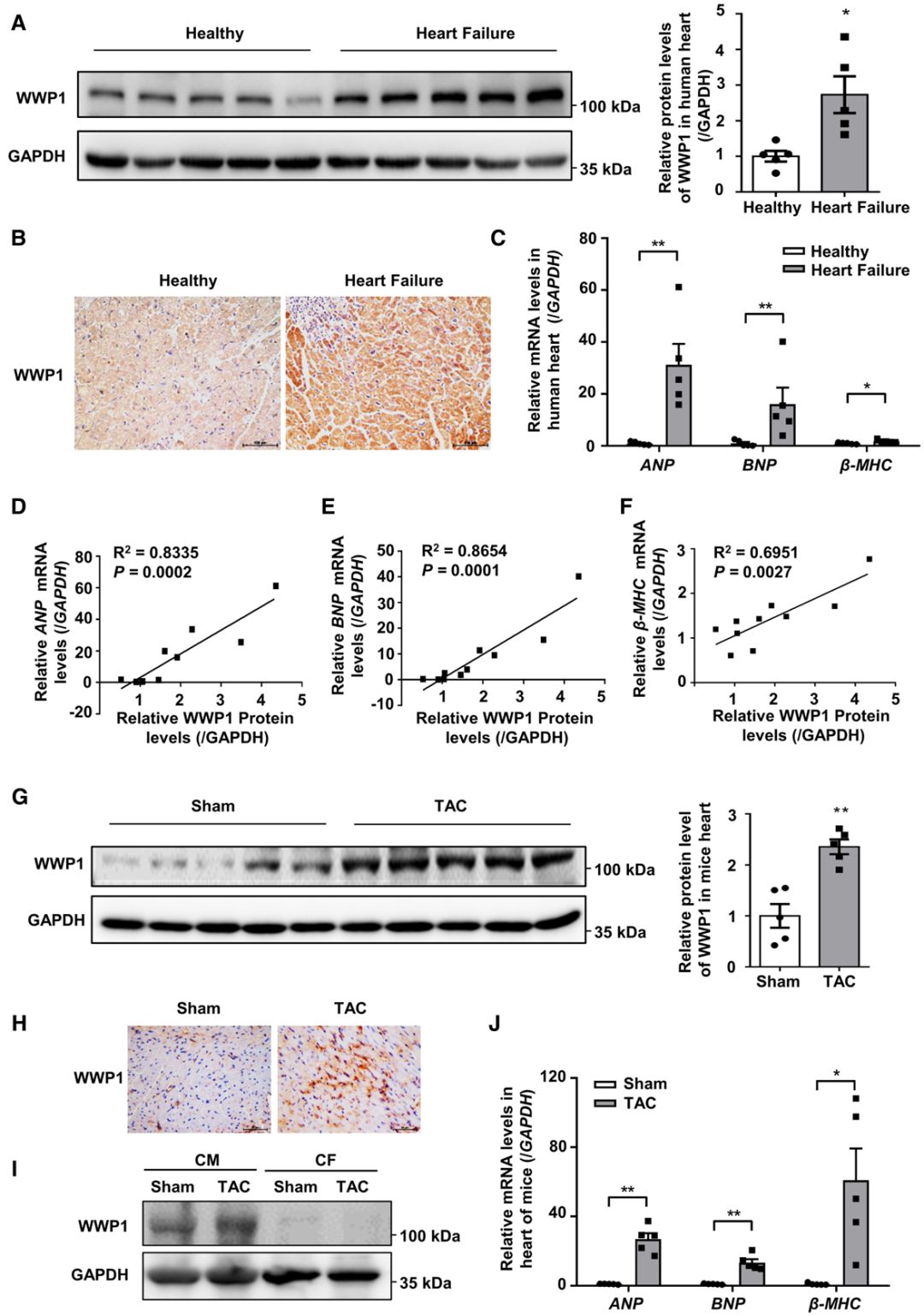
为评估WWP1在病理性肥厚心肌中的表达情况,研究人员分析了HF患者心脏组织中WWP1及肥厚标志物基因——心房利钠肽(ANP)、脑利钠肽(BNP)和β-肌球蛋白重链(β-MHC)的表达水平。蛋白质印迹法和免疫组织化学法检测结果显示,衰竭心脏中WWP1的蛋白水平显著高于正常对照组织。实时定量聚合酶链式反应(qPCR)结果表明,HF患者心脏组织中ANP、BNP和β-MHC的mRNA水平显著升高。进一步分析了WWP1与这3种HF标志物mRNA的相关性,结果与预期一致:WWP1与这些基因的表达水平均呈显著正相关。
为验证小鼠体内WWP1的表达特征,研究人员通过TAC构建病理性心脏肥厚模型,并采用蛋白质印迹法和免疫组织化学染色法分析WWP1的表达。结果显示,TAC术后4周,小鼠心脏中WWP1的蛋白水平显著升高;此外,从TAC术后成年小鼠体内分离的心肌细胞(CM)中WWP1蛋白水平升高,而心脏成纤维细胞(CF)中WWP1蛋白水平无明显变化。qPCR结果显示,TAC模型小鼠心脏中ANP、BNP和β-MHC的mRNA水平显著升高。这些结果表明,WWP1可能参与心脏重构的调控过程。
图1.在心肌肥厚和HF期间,人类和小鼠体内的WWP1蛋白水平升高
2.WWP1基因缺失对压力超负荷诱导的心肌肥厚具有保护作用
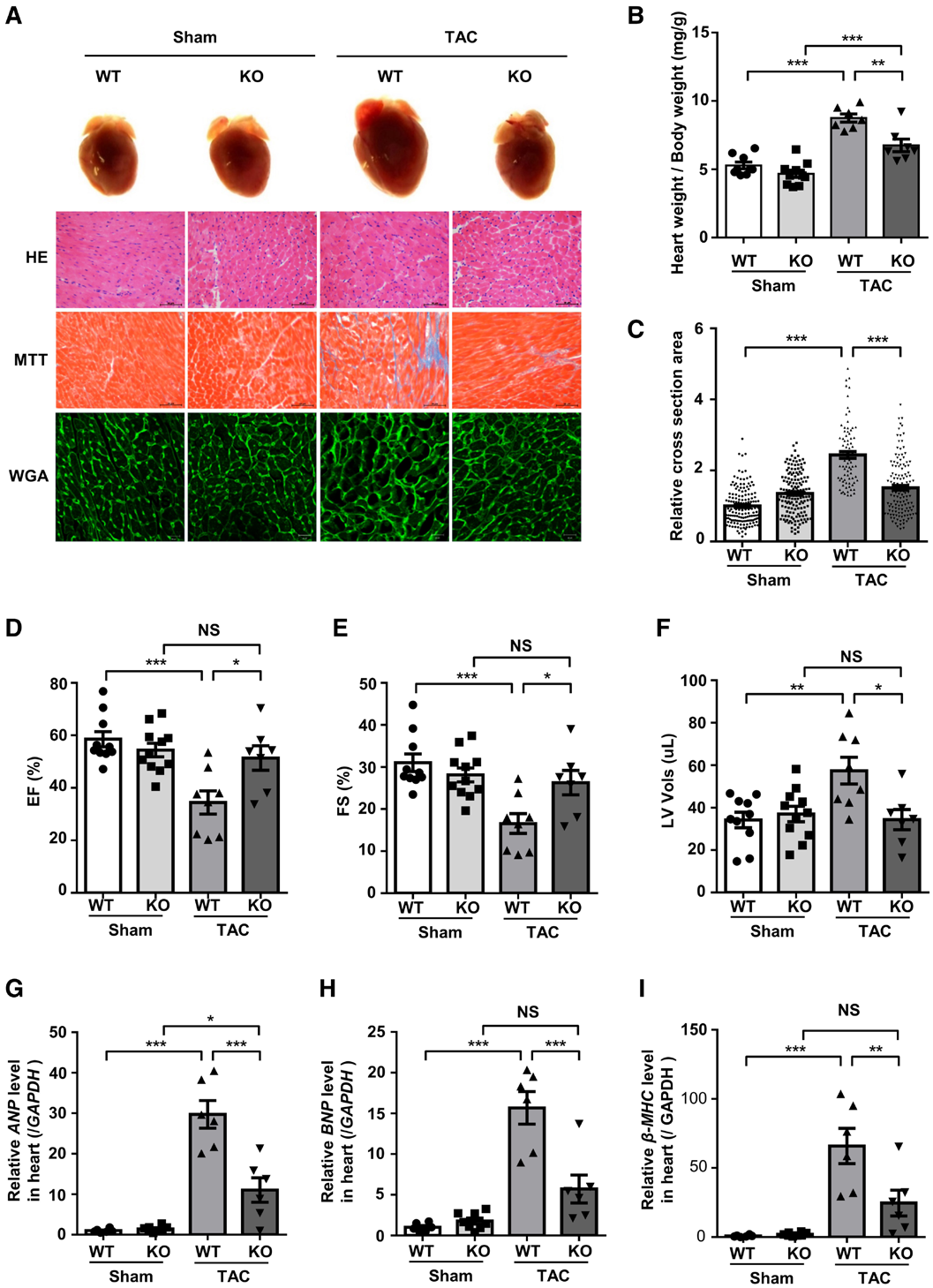
为进一步探究WWP1在心肌肥厚中的潜在作用,研究者对比了野生型(WT)小鼠与WWP1 KO小鼠在TAC术后的反应。结果显示,WWP1 KO小鼠的心脏中未检测到WWP1蛋白,其心肌细胞中也未检测到该蛋白。正如预期所示,WT小鼠出现明显心肌肥厚,表现为心肌细胞大小增加[麦胚凝集素(WGA)染色标记细胞边界,定量心肌细胞相对大小,即横截面积]、心脏重量/体重比升高及大面积纤维化。Masson三色染色(MT)结果显示,WWP1 KO小鼠心脏的纤维化程度更低。此外,WWP1 KO小鼠心脏重量/体重比及心肌细胞相对大小均显著降低。总的来说,与WT小鼠相比,WWP1 KO小鼠对TAC诱导的心肌肥厚反应更弱。
超声心动图结果显示,WT小鼠在TAC术后4周,射血分数(EF)和短轴缩短率(FS)显著下降,而WWP1 KO小鼠则未出现上述明显变化。此外,WT小鼠在TAC术后4周,左心室收缩末期容积、左心室收缩末期内径及左心室质量均显著增加;但WWP1 KO小鼠的上述指标无明显变化。
在基因表达方面,WWP1 KO小鼠心脏中,胎儿基因ANP、BNP及β-MHC的重新激活程度也显著低于WT小鼠心脏。综上,WWP1基因缺失可保护心脏免受压力超负荷诱导的心肌肥厚损伤。
图2.WWP1 KO对抗TAC诱导的心肌肥厚、胎儿基因激活及心肌纤维化
3.WWP1与DVL2存在相互作用
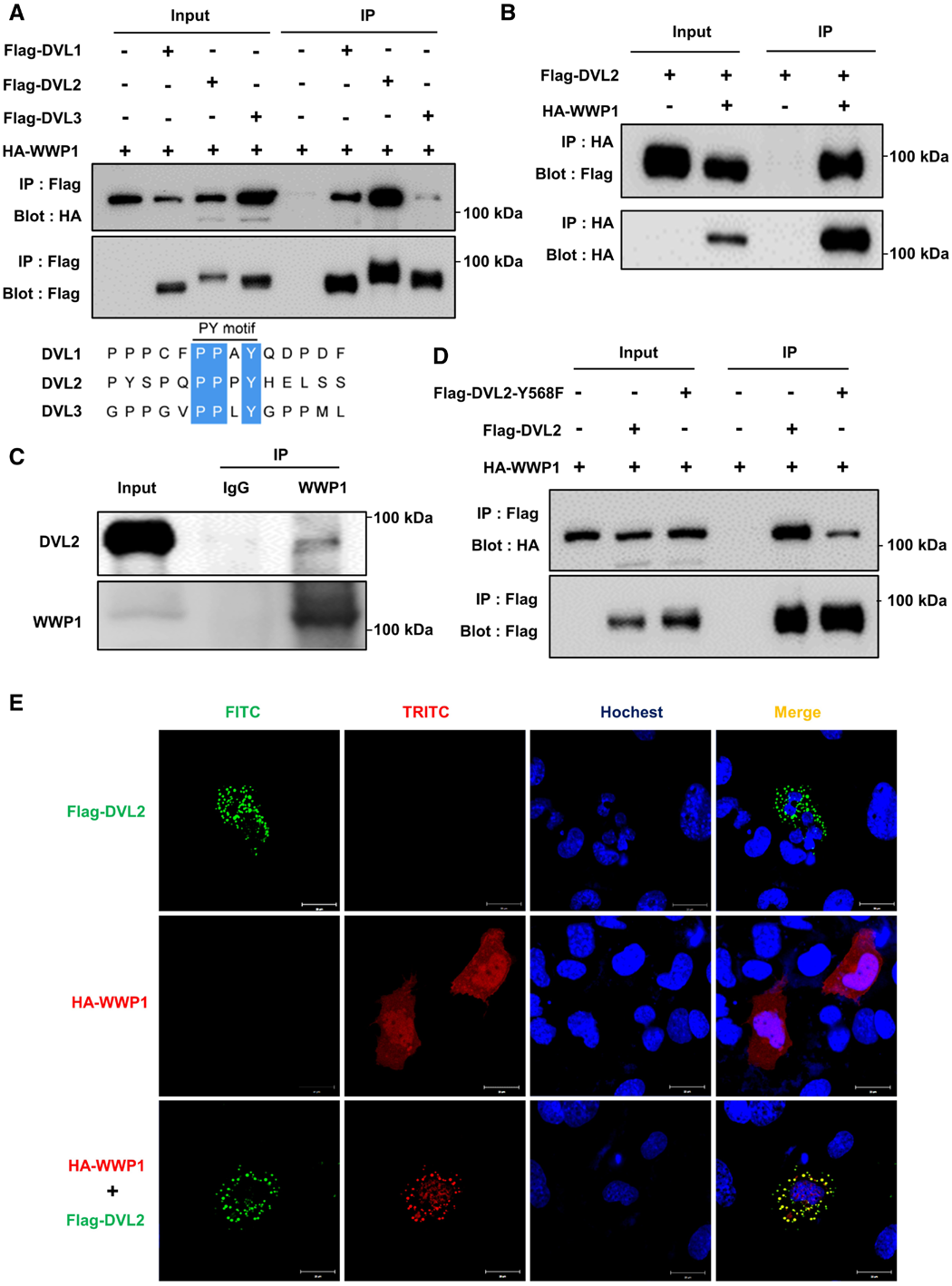
为探究与WWP1相互作用的蛋白,作者通过质谱分析鉴定了与WWP1共纯化的蛋白。结果显示,这些共纯化蛋白中包含DVL1和DVL2——而已知这两种蛋白是心脏重构的关键调控因子。为验证WWP1是否通过直接相互作用调控DVL蛋白功能,研究者在293T细胞中开展了免疫共沉淀实验。将Flag标签标记的DVL(Flag-DVL1、Flag-DVL2、Flag-DVL3),分别与血凝素(HA)标签标记的WWP1(HA-WWP1)共转染至293T细胞。结果显示,在Flag-DVL1、Flag-DVL2、Flag-DVL3组的免疫复合物中均检测到HA-WWP1;且Flag-DVL2组免疫沉淀样本中,WWP1的条带信号更强。因此,后续研究聚焦于DVL2。研究人员将Flag-DVL2与HA-WWP1共转染至293T细胞,结果显示,免疫复合物中检测到Flag-DVL2。此外,在293T细胞中,免疫共沉淀实验证实WWP1可与DVL2发生内源性结合,且对照免疫球蛋白G(IgG,同型对照)组未检测到DVL2的非特异性共沉淀。综上,WWP1可与DVL2直接相互作用。
由于DVL2的C端(羧基端)含有一个PY基序(序列为PPXY),且该基序在人类3种DVL蛋白中高度保守,研究人员进一步探究该PY基序是否通过物理相互作用被含WW结构域的WWP1识别。将突变型DVL2(Flag-DVL2-Y568F,即PY基序中酪氨酸Y突变为苯丙氨酸F)或野生型DVL2,分别与HA-WWP1共同转染293T细胞。结果显示,与Flag-DVL2组相比,Flag-DVL2-Y568F组免疫复合物中HA-WWP1的水平显著降低,提示DVL2的PY基序在其与WWP1的相互作用中发挥关键作用。
为明确WWP1与DVL2的空间定位关系,作者将Flag-DVL2与HA-WWP1分别或共同转染至小鼠心肌细胞(HL-1细胞)。共聚焦显微镜观察显示,WWP1与DVL2在HL-1细胞的细胞质中存在共定位。进一步验证了WWP1与DVL2存在相互作用。
接下来,研究者证实WWP1通过介导K27连接泛素化,维持DVL2的蛋白稳定性。
图3.DVL2可与WWP1发生相互作用
4.DVL2在人类衰竭心脏中与WWP1呈正相关,并介导WWP1对CaMKII-HDAC4-MEF2C信号轴的调控
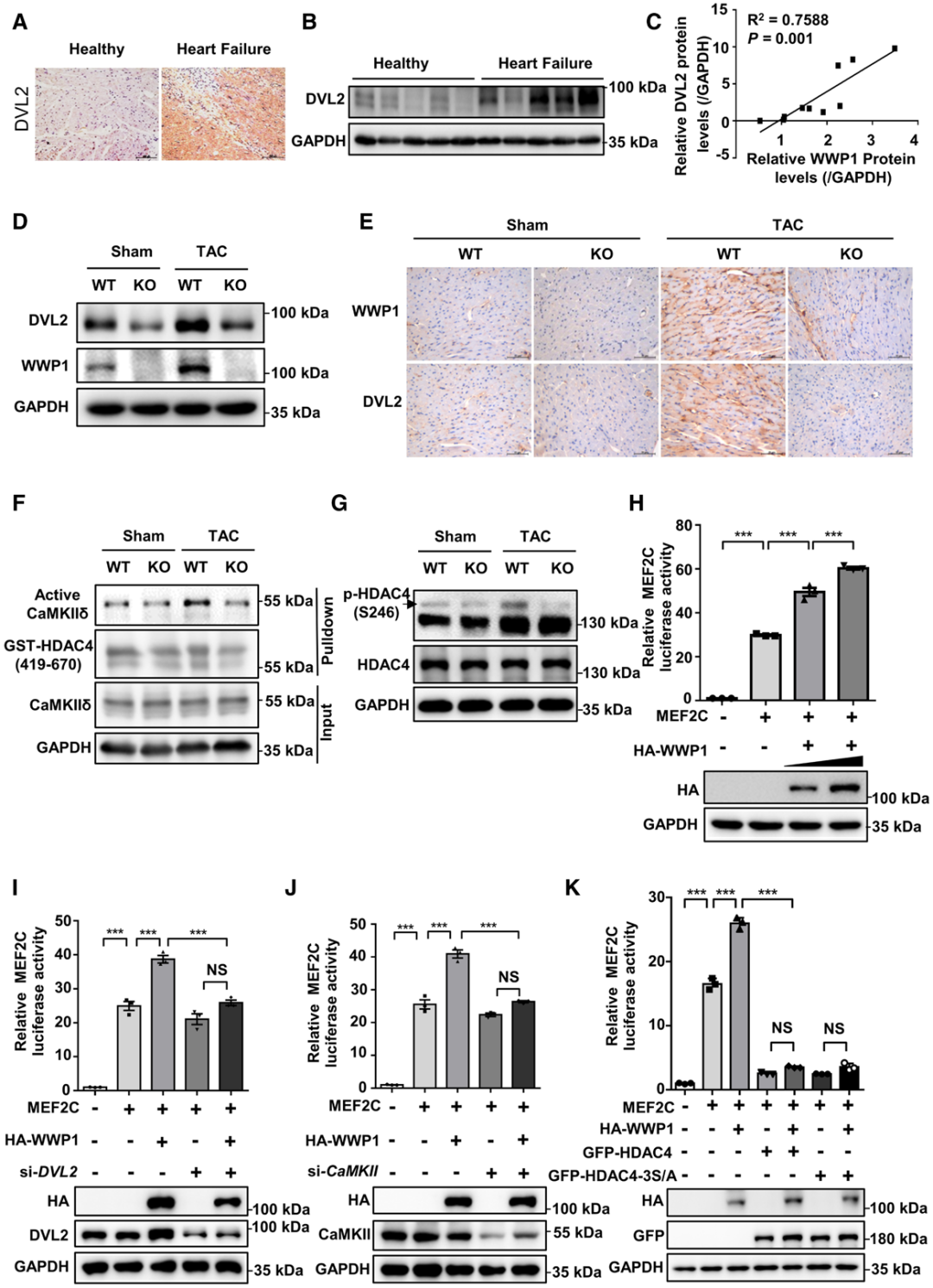
为在体内验证上述机制,研究人员分析了HF患者以及接受TAC的小鼠心脏中DVL2的蛋白水平。结果显示,HF患者心脏中DVL2蛋白水平显著升高,且与WWP1的表达呈显著正相关;而DVL1和DVL3的蛋白水平在HF患者心脏中无明显变化。在接受TAC的小鼠心脏中,DVL2蛋白水平同样显著升高,且这一效应可被WWP1缺失所抑制。此外,DVL2的mRNA水平未发生改变。因此,在心肌肥厚和HF过程中,WWP1与DVL2的蛋白稳定性密切相关。
为探究参与心脏重构调控的下游通路,作者筛选了可能受DVL2调控的心脏重构关键调控因子。DVL2是参与经典及非经典Wnt信号通路的重要胞质介导分子。于是首先检测了经典Wnt信号通路下游的关键信号分子及靶基因:有趣的是,在接受TAC的WWP1 KO小鼠与WT对照小鼠之间,活性形式β-连环蛋白(β-catenin)的蛋白水平,以及其靶基因(如Axin2、Nkd1、CyclinD1)的表达均无显著差异。随后,将研究重点转向非经典Wnt信号通路在心肌肥厚中的作用。CaMKII是非经典Wnt信号通路的重要调控因子,其持续激活形式在心脏重构和HF中起关键作用;近期有研究报道,CaMKII可与DVL2发生相互作用。已有研究表明DVL1可激活CaMKII,CaMKII激活后可直接磷酸化HDAC4,导致HDAC4向细胞质重新定位,进而激活MEF2C。实验结果显示,在接受TAC的WT小鼠心脏中,CaMKII的主要心脏亚型——活性形式CaMKIIδ的水平显著升高,而在WWP1 KO小鼠心脏中则无此变化;HDAC4的磷酸化水平变化趋势与CaMKII一致。此外,在HL-1细胞中,DVL2可与CaMKIIδ发生相互作用;进一步研究发现,心肌细胞特异性过表达DVL2提高了心脏中CaMKIIδ的活性,而敲低DVL2则降低了CaMKIIδ的活性。
为进一步验证WWP1对DVL2/CaMKII/HDAC4/MEF2C信号通路的影响,向293T细胞中转染了3×MEF2C结合位点荧光素酶报告基因质粒、MEF2C表达质粒,同时加入或不加入WWP1表达质粒。结果显示,在293T细胞中,WWP1可通过剂量依赖性方式增强MEF2C荧光素酶报告基因的活性。而当加入DVL2小干扰RNA(siRNA)、CaMKII siRNA、HDAC4表达载体或HDAC4 3S/A[组成型核定位型:3个关键的丝氨酸(Ser)残基突变为丙氨酸(Ala),从而使其丧失磷酸化能力,导致HDAC4被“锁定”在细胞核内,无法通过磷酸化依赖的机制转运到细胞质中]载体后,WWP1对MEF2C活性的增强效应显著减弱;综上,这些结果表明:在人类HF中,DVL2与WWP1呈正相关,且DVL2可介导WWP1对CaMKII-HDAC4-MEF2C信号轴的调控作用。
图4.在人类衰竭心脏中,DVL2与WWP1呈正相关,且介导WWP1对CaMKII-HDAC4-MEF2C通路的调控作用
5.DVL2/CaMKII信号通路在WWP1调控心肌肥厚中起关键作用
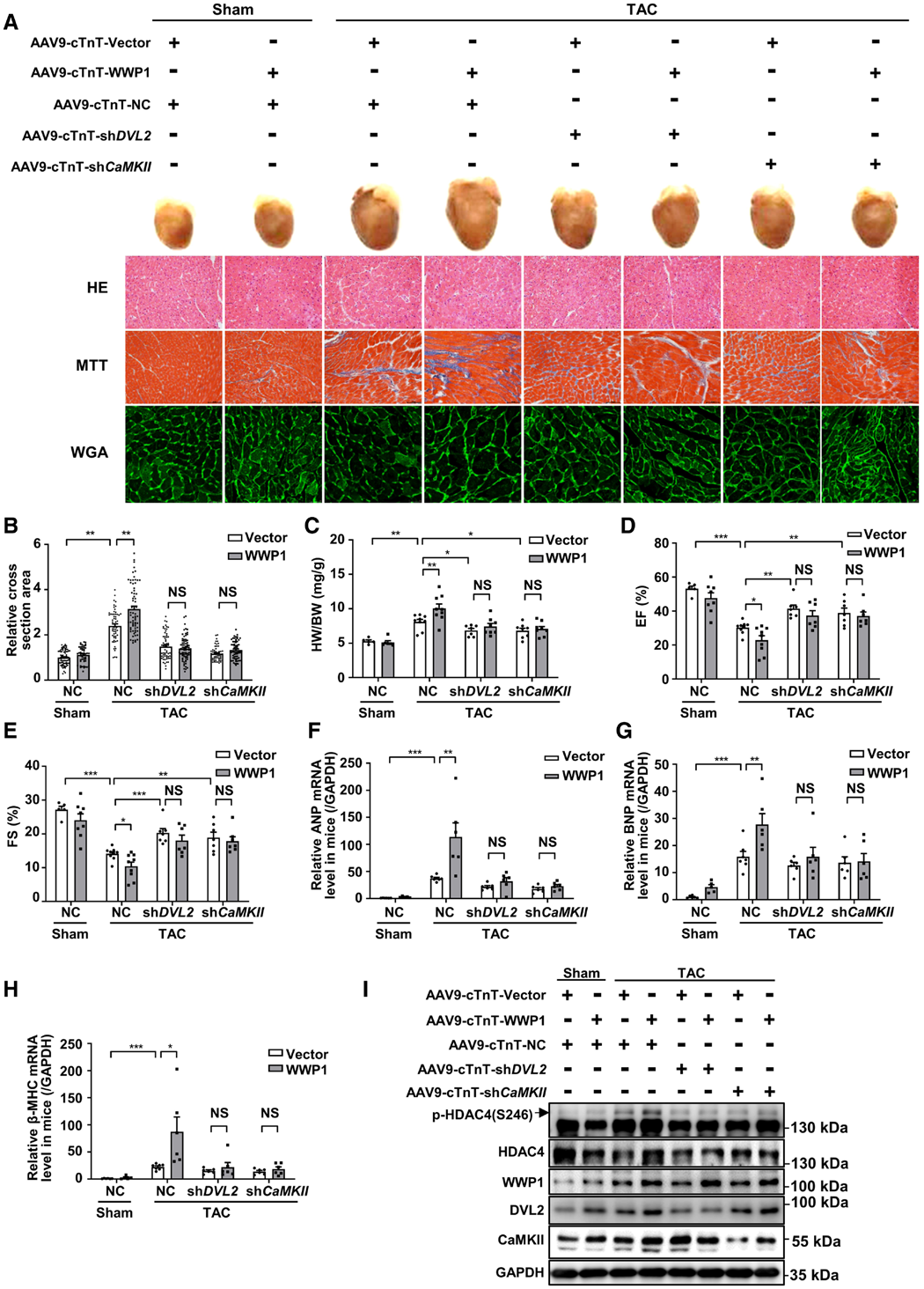
为验证DVL2/CaMKII信号通路在WWP1调控心肌肥厚中的作用,研究人员构建了受心肌细胞特异性cTnT启动子调控的AAV9病毒载体,用于在小鼠中过表达WWP1,以及敲低DVL2和CaMKII。在TAC术后1周,给小鼠尾静脉注射AAV9-cTnT-WWP1以过表达WWP1,同时部分小鼠联合注射AAV9-cTnT-shDVL2或AAV9-cTnT-shCaMKIIδ,继续观察3周。正如预期,TAC术后心肌细胞特异性过表达WWP1的小鼠出现明显心肌肥厚,表现为心肌细胞大小增加、心脏重量/体重比升高以及大面积纤维化。而心肌细胞特异性敲低DVL2或CaMKII逆转了WWP1对心肌细胞大小和心肌纤维化的影响。
超声心动图分析显示,与空载组相比,TAC术后4周WWP1过表达小鼠的EF和FS显著降低,而敲低DVL2或CaMKII逆转了这一趋势。心肌细胞特异性过表达WWP1的小鼠心脏中,胎儿基因ANP、BNP和β-MHC的表达也升高,而这一效应被DVL2或CaMKII敲低明显抑制。此外,心肌细胞特异性过表达WWP1升高了心脏中磷酸化HDAC4(p-HDAC4)的水平,但敲低DVL2或CaMKII逆转了这一变化。体内实验还显示,敲低DVL2减弱了WWP1对CaMKIIδ活性的调控。综合来看,心肌细胞特异性敲低DVL2或CaMKII显著抑制了WWP1对心肌肥厚的促进作用。
研究者还构建了受心肌细胞特异性cTnT启动子调控的AAV9病毒载体,用于在心肌细胞中过表达DVL2。在TAC术后1周,给WWP1 KO小鼠注射AAV9-cTnT-DVL2以过表达DVL2,继续观察3周。组织学和超声心动图结果显示,心肌细胞特异性过表达DVL2阻断了WWP1 KO对心肌纤维化、心肌细胞大小以及心功能的保护作用。在WWP1 KO小鼠的心脏中,TAC术后过表达DVL2,胎儿基因ANP、BNP和β-MHC的重新激活也得以恢复。此外,WT小鼠中,TAC诱导p-HDAC4水平升高;而在WWP1 KO小鼠中,TAC无法有效诱导p-HDAC4升高,当在其心脏中过表达DVL2后,p-HDAC4水平才得以恢复至WT小鼠TAC术后的升高水平。因此,DVL2/CaMKII信号通路在WWP1调控心肌肥厚中起关键作用。
图5.DVL2/CaMKII信号通路对WWP1调控心肌肥厚的过程至关重要
6.靶向WWP1可对抗压力超负荷诱导的心肌肥厚
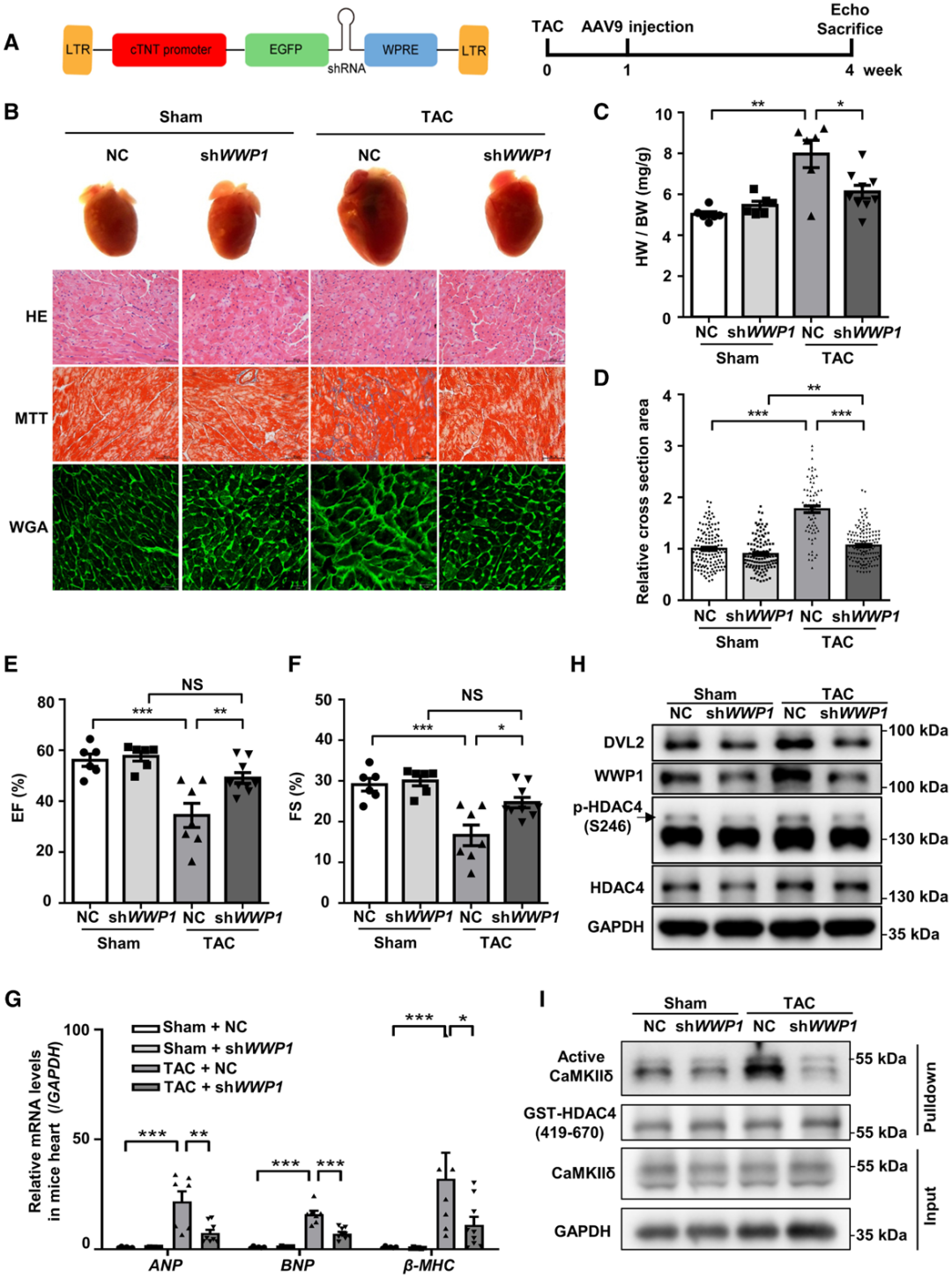
为评估WWP1在HF治疗中的潜力,作者对WT小鼠实施TAC以诱导心肌肥厚。同时,构建了一种受cTnT启动子调控的shRNA载体,用于在心肌细胞中敲低WWP1的表达。在TAC术后1周,向小鼠注射AAV9-cTnT-shWWP1,以敲低内源性WWP1的表达,继续观察3周。正如预期,注射AAV9-cTnT-NC的小鼠出现明显的心肌肥厚,表现为心肌细胞大小增加、心脏重量/体重比升高以及大面积纤维化。与注射AAV9-cTnT-NC的小鼠相比,注射AAV9-cTnT-shWWP1的小鼠对TAC诱导的心肌肥厚反应减弱:TAC术后,其心脏重量/体重比及心肌细胞相对大小均显著降低。MT结果显示,注射AAV9-cTnT-shWWP1的小鼠心脏纤维化程度更轻。
超声心动图分析显示,TAC术后4周,注射AAV9-cTnT-NC的小鼠EF和FS显著下降,而注射AAV9-cTnT-shWWP1的小鼠未出现此类明显变化。在AAV9-cTnT-NC小鼠中,TAC术后4周导致左心室质量增加、左心室舒张末期后壁厚度增厚及左心室收缩末期容积增大;然而,注射AAV9-cTnT-shWWP1的小鼠中该反应显著减弱。与AAV9-cTnT-NC小鼠的心脏相比,AAV9-cTnT-shWWP1小鼠心脏中胎儿基因(ANP、BNP、β-MHC)的重新激活程度降低。与注射AAV9-cTnT-NC相比,注射AAV9-cTnT-shWWP1显著降低了心脏中WWP1蛋白的表达水平。此外,TAC术后升高的DVL2、活化CaMKIIδ及p-HDAC4水平,在注射AAV9-cTnT-shWWP1后均恢复正常,这与WWP1 KO的实验结果一致。这些结果表明,在心肌细胞中特异性敲低WWP1可保护心脏免受压力超负荷诱导的心肌肥厚影响,且WWP1有望成为心肌肥厚及HF的治疗靶点。
图6.以WWP1为治疗靶点,可通过抑制DVL2-CaMKII-HDAC4-MEF2C通路,对抗压力超负荷诱导的心肌肥厚
总结
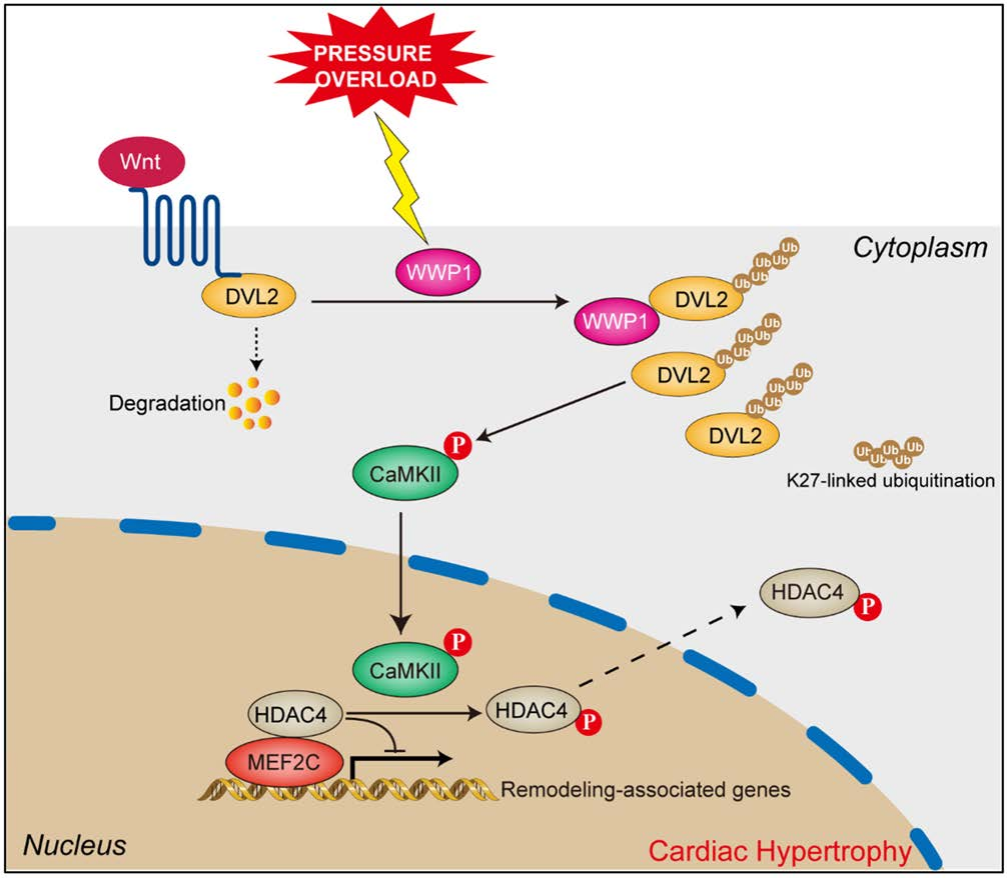
本研究证实,WWP1是病理性心肌肥厚及HF的新型调控因子。在HF患者及TAC术后小鼠的肥厚心脏中,WWP1水平均显著升高。在WWP1 KO小鼠中,压力超负荷诱导的心肌肥厚程度得到显著缓解。机制层面,WWP1可通过介导DVL2的K27连接多聚泛素化维持其蛋白稳定性,进而通过DVL2/CaMKII/HDAC4/MEF2C通路调控心脏重构。此外,利用AAV9-cTnT-shWWP1对WWP1进行治疗性靶向干预,几乎完全消除了TAC诱导的心肌肥厚及心功能损伤等不利影响。上述结果共同提示,WWP1可能是治疗病理性心肌肥厚及HF的潜在靶点。
模式图:WWP1在压力超负荷诱导的心肌肥厚中的功能机制
本文使用的病毒产品,列表如下:


了解产品及服务
扫码添加客服微信:BrainVTA2020

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK