2025-09-01 阅读量:1458
全球糖尿病患病率正快速上升,预计到2045年,糖尿病将影响全球10.9%的人口。2型糖尿病患者会发生糖尿病心肌病(DCM),这是一种以心肌结构与功能异常为特征的疾病表型,由糖尿病相关炎症、糖脂代谢紊乱及线粒体损伤引起,且与高血压、冠状动脉疾病及心脏瓣膜病无关。因此,针对糖尿病心脏代谢紊乱的有效干预策略,有望为DCM提供新型治疗选择。
线粒体功能障碍在DCM发病机制中发挥关键作用,其与线粒体动力学(包括线粒体分裂与融合)密切相关。线粒体分裂由线粒体动力相关蛋白调控,包括Fis1(线粒体分裂蛋白1)、Drp1(动力相关蛋白1)及Mff(线粒体分裂因子);而线粒体融合主要由Mfn1(线粒体融合蛋白1)、Mfn2(线粒体融合蛋白2)及Opa1(视神经萎缩蛋白1)调控。线粒体过度分裂被认为具有致病性:过度分裂会增加线粒体数量但减小其体积,导致大量线粒体断裂、活性氧(ROS)大量生成,并加剧线粒体功能障碍。另一方面,Mfn2可通过促进线粒体融合改善线粒体功能障碍,从而延缓病理性心脏病变进展。值得一提的是,有研究发现,心脏缺失Mfn2后,小鼠在缺血损伤后反而呈现出一定的心脏保护作用。但总体而言,线粒体融合蛋白表达或功能降低,通常会干扰线粒体功能的核心机制,进而可能诱发心脏病变,在糖尿病心脏中尤为明显。既往研究已证实,Mfn2缺失会破坏内质网与线粒体间的相互作用,损害线粒体钙(Ca2+)摄取功能,并对心肌细胞收缩功能产生不利影响;此外,Mfn2缺失还会破坏自噬小体-溶酶体融合过程,逐步导致心功能障碍。因此,提高心肌细胞中Mfn2的表达,或可为DCM提供潜在治疗方向。
泛素化是一种蛋白质翻译后修饰过程,在此过程中,泛素小分子标记蛋白质,使其被蛋白酶体介导降解。泛素化在调控细胞内蛋白质稳态及酶活性中发挥核心作用。去泛素化酶可通过从多肽链上移除泛素分子来拮抗E3泛素连接酶活性,是泛素化调控过程中的关键分子。目前已在人类中鉴定出约100种去泛素化酶,主要分为两大类:半胱氨酸蛋白酶和金属蛋白酶。其中,半胱氨酸蛋白酶包括UCH(泛素羧基末端水解酶)家族、USP(泛素特异性蛋白酶)家族、OTU(卵巢肿瘤样蛋白酶)家族及马查多-雅各布病蛋白酶。USP28(泛素特异性蛋白酶28)作为USP家族成员,参与多种生理过程,如DNA损伤修复、细胞凋亡、细胞增殖及分化。致癌基因c-MYC是E3泛素连接酶的重要底物,而USP28可拮抗E3泛素连接酶活性、稳定致癌基因c-MYC,从而促进肿瘤细胞增殖、肿瘤发生及转移。此外,USP28还可通过抑制细胞凋亡、上调血管生成,推动肿瘤发展。既往关于USP28的研究主要集中于肿瘤信号通路,但其在心血管疾病(尤其是糖尿病心脏病变)中的作用尚不明确。
过氧化物酶体增殖物激活受体(PPARs)包括PPARα、PPARδ及PPARγ三种亚型,是调控糖脂代谢及能量稳态相关基因表达的转录因子。PPARα作为一种脂肪酸(FA)感受器,在心脏中呈较高水平表达,其激活可促进线粒体FA氧化。糖尿病小鼠心肌组织中PPARα含量降低,且线粒体FA氧化相关基因表达显著下调。衰竭心脏的PPARα表达同样降低,且能量供应偏好葡萄糖、酮体及乳酸,而非FA,这会导致线粒体FA氧化相应减少、脂质代谢紊乱,进而引起心肌收缩性下降及心功能恶化。需要特别留意的是,有研究提示,心脏PPARα异常高表达可能是DCM发生发展的重要因素,尤其在2型糖尿病或1型糖尿病小鼠疾病早期至中期(6-15周)——此阶段小鼠会出现轻至中度心肌舒张功能障碍。然而,人类研究显示,在伴有严重舒张功能障碍的2型糖尿病患者心脏中,PPARα表达未发生显著改变或仅轻度降低。因此,PPARα在DCM心功能障碍(长期糖尿病状态下的心功能障碍)发生发展中的作用评估尚不充分,仍需进一步研究。
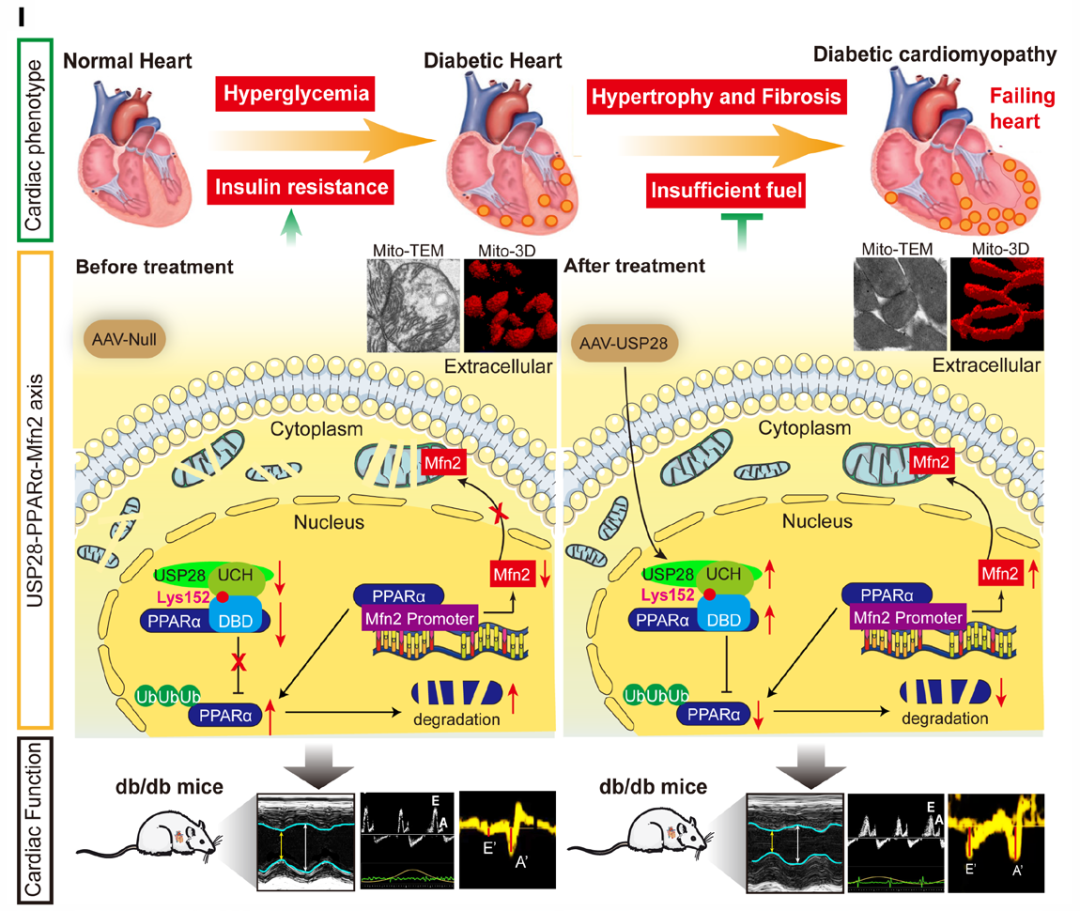
近期,武汉大学人民医院唐其柱主任医师团队在Circulation期刊(IF=37.8)发表了以“USP28 Serves as a Key Suppressor of Mitochondrial Morphofunctional Defects and Cardiac Dysfunction in the Diabetic Heart”为题的研究论文。该研究证实,USP28可通过改善糖尿病小鼠心脏的收缩及舒张功能障碍、减轻心肌肥厚与纤维化,对长期糖尿病状态下的心脏发挥保护作用。此外,USP28还能显著抑制糖尿病小鼠心脏的线粒体形态和功能缺陷,并调控心肌脂质蓄积。机制层面,作者发现USP28的UCH结构域可直接与PPARα的DNA结合结构域(DBD)结合,进而促进PPARα第152位赖氨酸位点的去泛素化,稳定PPARα蛋白并促进心肌细胞中Mfn2的转录,最终增强线粒体融合、改善线粒体功能障碍。该研究结果揭示了糖尿病心脏中一种由USP28调控、涉及PPARα-Mfn2轴的线粒体稳态机制,提示USP28激活或腺相关病毒(AAV)靶向治疗或许能成为DCM的潜在治疗策略。
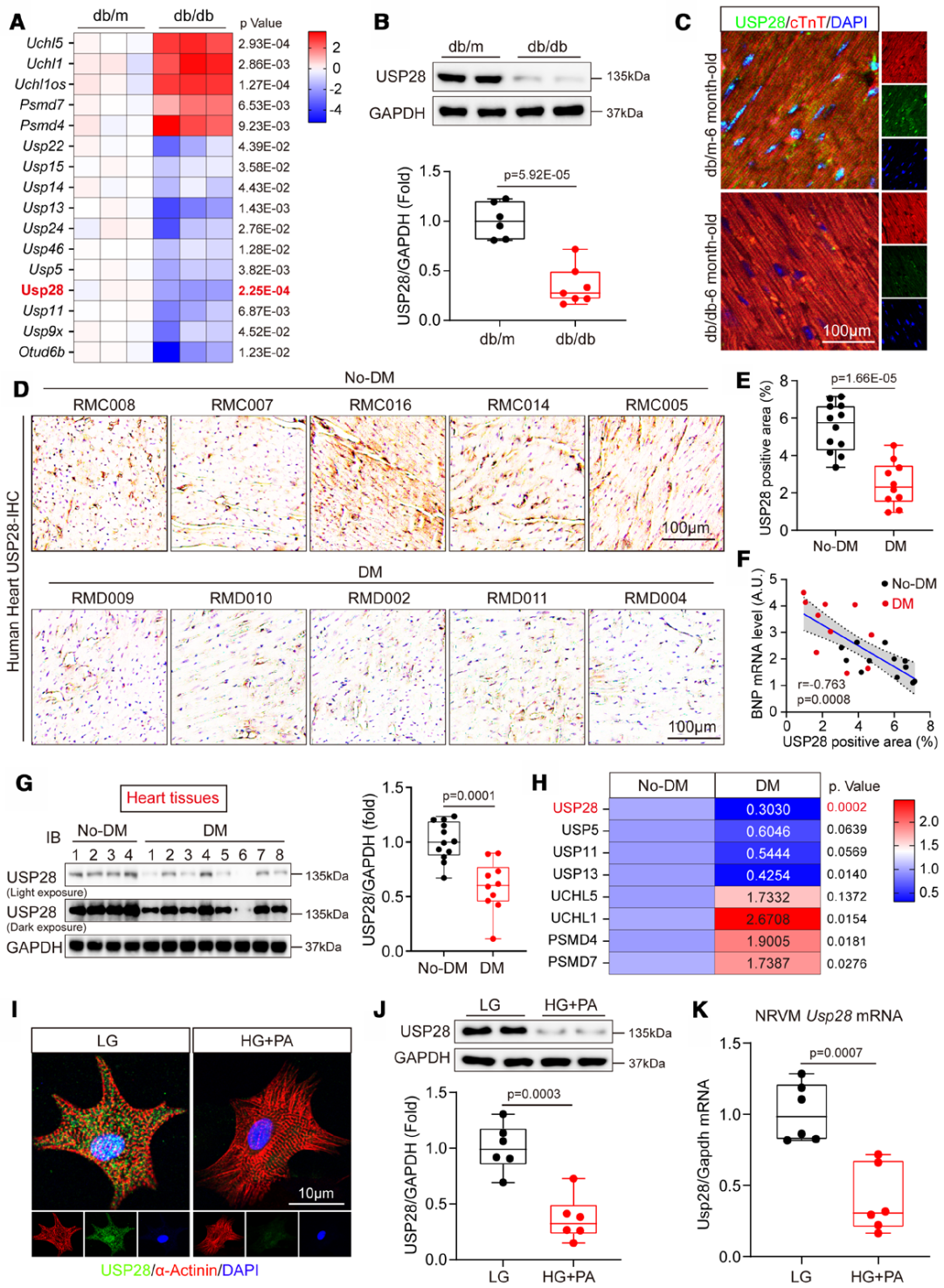
1.糖尿病心脏中USP28表达显著下调,且与DCM进展相关
为筛选糖尿病诱导心功能障碍的潜在关键调控因子,研究人员分析了db/db小鼠(瘦素受体缺陷,自发2型糖尿病模型小鼠,通过db/m小鼠自交获得)与db/m小鼠(正常对照小鼠)左心室组织的基因表达谱。对差异表达基因(DEGs)的基因本体(GO)分析显示,泛素介导的蛋白水解过程显著富集。为探究USP家族中具有功能意义的基因,进一步分析了与去泛素化酶相关的DEGs,共鉴定出5个上调基因与11个下调基因,其中USP28在糖尿病心脏中的下调最为显著。此外,既往研究数据显示USP28在人类心脏组织中高度富集,且db/db小鼠左心室组织中USP28 mRNA水平显著降低,尤其是心肌细胞中的USP28表达量,与对照组相比下降更为明显。因此,作者在后续实验中聚焦于USP28的功能研究。首先,通过蛋白质印迹与免疫荧光染色验证,糖尿病小鼠心脏中USP28表达确实降低。
为探究USP28在正常心脏向糖尿病心脏转变过程中的作用,研究者采用免疫组织化学染色,检测了健康供体与糖尿病患者代表性心脏样本中的USP28表达。结果显示,糖尿病患者心脏组织中USP28蛋白水平显著降低。这里要注意的是,心脏USP28表达水平与B型利钠肽(BNP)所评估的心力衰竭严重程度呈负相关。同样,蛋白质印迹与实时聚合酶链反应结果显示,糖尿病患者心脏中USP28的mRNA与蛋白水平均显著低于非糖尿病个体。
随后,作者用高糖联合棕榈酸(HG+PA)处理新生大鼠心室肌细胞(NRVMs)24小时,以模拟体内高糖高脂环境。与在人类和db/db小鼠中的发现一致,相较于低糖(LG)处理组,HG+PA处理组NRVMs的USP28表达显著降低。为进一步验证这一结论,在体外实验中使用了人诱导多能干细胞分化心肌细胞(hiPSC-CMs),结果显示,经HG+PA处理后,hiPSC-CMs中的USP28表达同样降低。
尤为关键的是,在2型糖尿病刺激下,尽管细胞质中USP28表达仅轻微下降,但细胞核中USP28表达量显著降低,提示核定位USP28可能与2型糖尿病诱导的心脏表型改变密切相关。USP28表达与DCM的这一显著相关性,揭示了USP28在DCM疾病进展中的作用。
图1.USP28在糖尿病心脏中的表达显著下调,并与DCM的进展相关
图2.db/m或db/db小鼠心脏细胞质和细胞核成分中USP28蛋白表达
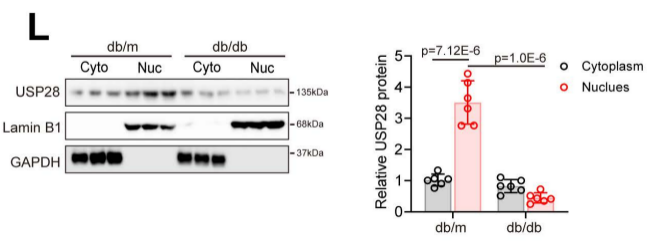
2.恢复心脏USP28可改善糖尿病心脏的重构与功能障碍
为明确USP28在糖尿病心脏中的作用,研究人员通过尾静脉注射,向6周龄db/db小鼠体内递送了心肌靶向性重组腺相关病毒9型(rAAV9-cTnT-USP28-3×Flag-P2A-GFP,简称rAAV9-USP28),以实现心肌细胞特异性过表达USP28;同时以空载病毒(rAAV9-null)处理的小鼠作为对照。随后,所有小鼠均饲养至24周龄,并在该时间点接受超声心动图检查。
通过蛋白质印迹以及Flag标签的免疫荧光染色(同时直接观察绿色荧光),验证了心肌组织中USP28的过表达效率。进一步的蛋白质印迹结果显示,注射rAAV9-USP28显著提高了小鼠心脏中USP28蛋白的表达水平,但对肝脏、肾脏中USP28的表达无影响。在空腹血糖(FBG)、体重、血压及心率方面,恢复USP28表达的db/db小鼠与未恢复USP28的db/db小鼠之间无显著差异。
接下来,通过超声心动图评估小鼠心功能,发现db/db小鼠存在明显的左心室收缩功能障碍;而注射rAAV9-USP28可部分改善该障碍,具体表现为左心室射血分数(LVEF)、左心室短轴缩短率(LVFS)升高,且左心室质量降低。在舒张功能评估中,采用二尖瓣舒张早期血流速度与舒张晚期血流速度的比值(E/A比值)作为指标。多普勒超声心动图检测的二尖瓣流入速度显示,db/db小鼠的E/A比值显著升高(>2.5),提示舒张功能受损;与之相反,rAAV9-USP28处理降低了左心室舒张功能障碍程度、改善心脏舒张功能,具体证据为E/A比值下降。此外,采用二尖瓣舒张早期血流速度与二尖瓣环舒张早期组织速度的比值(E/E'比值)评估左心室充盈压,并通过检测二尖瓣环处的组织多普勒信号计算舒张早期峰值速度(E')与心房收缩期峰值速度(A')。结果显示,24周龄db/db小鼠的E/E'比值显著升高;而心肌细胞中过表达USP28显著降低了db/db小鼠的E/E'比值。
为探究USP28对心肌细胞功能的影响,研究人员检测了24周龄rAAV9-USP28处理组与rAAV9-null处理组db/db小鼠离体心肌细胞的胞质钙瞬变。结果显示,与对照组相比,恢复USP28表达改善了心肌细胞的胞质钙瞬变。以上结果表明,恢复USP28改善了db/db小鼠的心脏收缩与舒张功能。
研究者还通过评估心脏形态,探究了USP28过表达对db/db小鼠心脏结构重构的影响。结果显示,USP28减轻了db/db小鼠的心肌肥厚,具体表现为心脏体积缩小、心脏重量/胫骨长度比值降低。同样,麦胚凝集素(WGA)染色结果显示,USP28过表达组db/db小鼠的心肌细胞横截面积小于rAAV9-null处理组db/db小鼠。此外,通过Masson染色检测心肌胶原体积分数发现,USP28减少了db/db小鼠左心室的胶原沉积。蛋白质印迹结果进一步证实了USP28在心脏重构中的保护作用,具体表现为BNP、β-肌球蛋白重链(β-MHC)及Ⅰ型胶原(CollagenⅠ)的蛋白水平降低。综上,这些数据表明,恢复心脏USP28的表达可改善糖尿病心脏的重构与功能障碍。
图3.心脏USP28的恢复可改善糖尿病心脏的重构和功能障碍
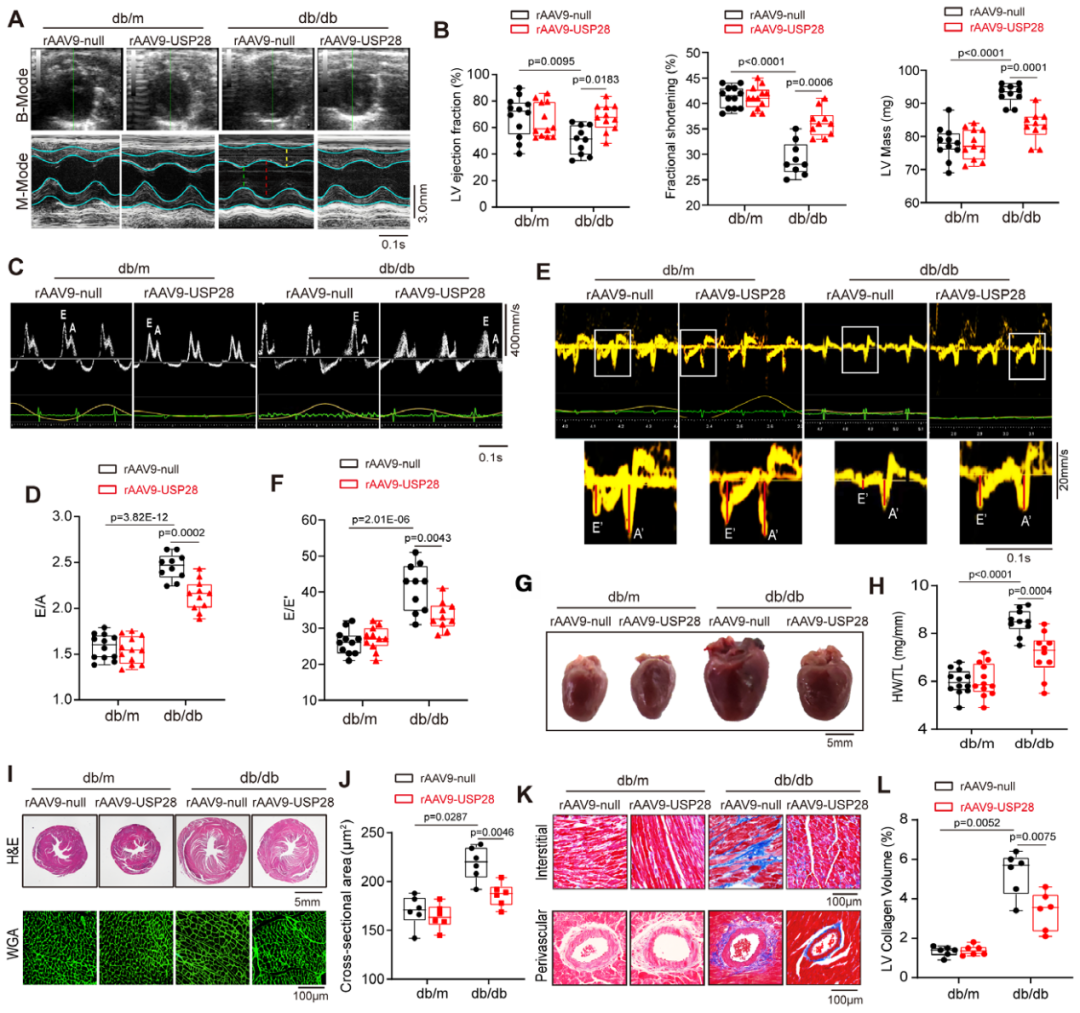
3.USP28可抑制脂质蓄积并逆转ROS生成及线粒体功能障碍
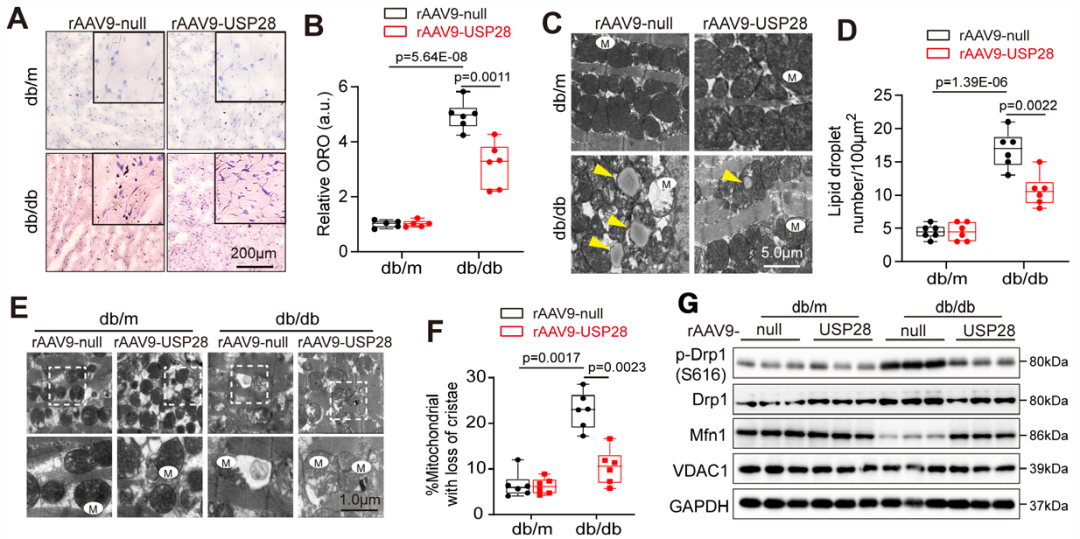
为探究USP28改善心脏重构与功能障碍的机制,作者对rAAV9-USP28处理组与rAAV9-null处理组db/db小鼠的左心室组织进行了全面的RNA测序分析。对两组糖尿病心脏DEGs的GO分析显示,代谢过程与线粒体过程相关基因显著富集。随后,通过检测心脏组织中的脂质含量与线粒体形态,探究USP28过表达在DCM中的作用。油红O染色结果显示,db/db小鼠心脏存在明显脂质蓄积;与rAAV9-null处理组db/db小鼠相比,USP28过表达显著减轻了这种脂质蓄积。脂质组学分析同样表明,糖尿病心脏存在脂质过载(神经酰胺Cer与二酰甘油DG水平升高),而恢复USP28表达缓解了该现象。此外,USP28还减少了db/db小鼠心肌中的脂滴数量。
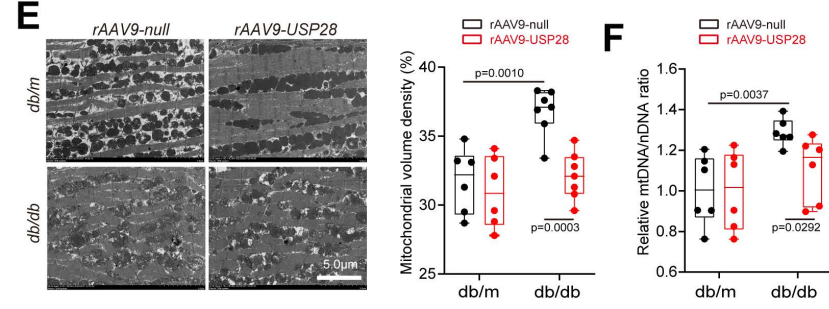
在24周龄db/db小鼠的心肌组织中,研究人员还观察到线粒体异常——线粒体嵴出现明显损伤,表现为嵴缺失与肿胀。USP28过表达对db/m小鼠的线粒体形态无影响,但显著降低了db/db小鼠心肌中嵴缺失线粒体的比例,并促进三磷酸腺苷(ATP)生成。随后,研究者通过蛋白质印迹检测线粒体融合与分裂相关关键蛋白的表达水平发现:相比于db/m小鼠,糖尿病心脏中Mfn1的表达显著下调,而注射rAAV9-USP28恢复了db/db小鼠心脏中Mfn1的表达;此外,USP28过表达还抑制了db/db小鼠心肌组织中Drp1的S616位点(Drp1S616)磷酸化——该位点磷酸化会增强应激状态下Drp1的功能,进而诱导线粒体分裂。与蛋白质检测结果一致,RNA测序结果同样显示:USP28过表达组小鼠的线粒体分裂相关基因(如Mff、动力相关蛋白2/Drp2)表达显著降低,而线粒体融合相关基因(如Bik样杀伤蛋白Blk、Mfn1、Mfn2、视神经萎缩蛋白3/Opa3)表达显著升高。同时,恢复USP28表达还促进了糖尿病心脏中FA氧化相关基因的表达,从而改善脂质代谢。
接下来,研究者通过电镜分析与线粒体DNA含量检测评估线粒体丰度,并通过蛋白质印迹检测氧化磷酸化(OXPHOS)复合物以评估线粒体完整性。已有研究证实,糖尿病心脏中线粒体融合受损且线粒体自噬受抑制,导致线粒体体积密度与线粒体DNA含量升高;本研究表明恢复USP28表达缓解了db/db小鼠的这些异常变化。此外,对OXPHOS复合物Ⅰ-Ⅴ主要亚基的蛋白质印迹分析显示,各组间无明显差异。
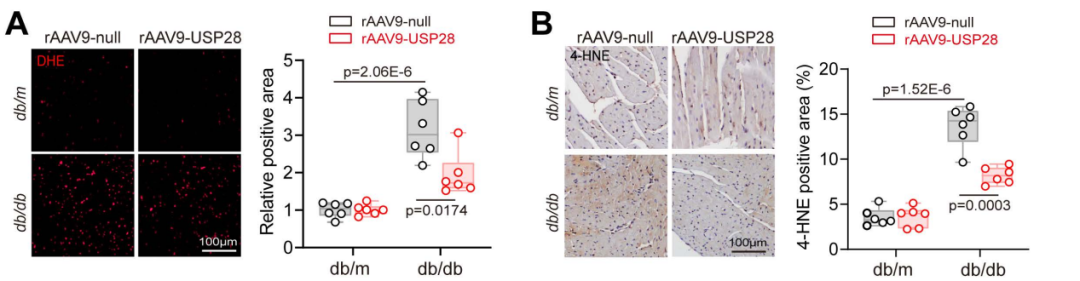
线粒体功能缺陷与脂质过氧化及ROS生成密切相关。因此,作者通过二氢乙锭(DHE)检测与4-羟基壬烯醛(4-HNE)免疫组织化学染色,分别评估心肌组织中的ROS水平与脂质过氧化程度。结果显示,db/db小鼠心脏的ROS含量与脂质过氧化水平显著升高,而USP28表达上调抑制了ROS蓄积。此外,既往癌症领域研究表明,USP28可通过上调血管生成促进癌症发展。因此,研究人员进一步探究USP28对糖尿病心脏血管生成的影响。结果符合预期:24周龄db/db小鼠存在血管生成障碍,而rAAV9-USP28处理恢复了其血管生成能力。总的来说,这些结果表明,恢复USP28表达显著改善了糖尿病诱导的心肌脂质蓄积与线粒体功能缺陷。
图4.USP28抑制db/db小鼠心肌组织中脂质积累并逆转线粒体功能障碍
图5.db/db小鼠心肌细胞中过表达USP28后,心肌线粒体体积密度与线粒体DNA含量的异常升高均得到有效降低
图6.糖尿病心脏中USP28抑制ROS的产生
进一步地,研究人员通过将Myh6-Cre+/USP28fl/fl小鼠与db/m小鼠进行杂交繁育,构建了心肌特异性USP28敲除db/db小鼠;体外实验中利用USP28小干扰RNA(siUSP28)实现USP28沉默,以探究心肌USP28在DCM发病机制中的作用。结果显示,心肌特异性USP28缺失加重了糖尿病性心力衰竭、心脏重构、脂质过载及线粒体功能障碍。
4.USP28通过与PPARα特异性相互作用,调控其去泛素化修饰及稳定性
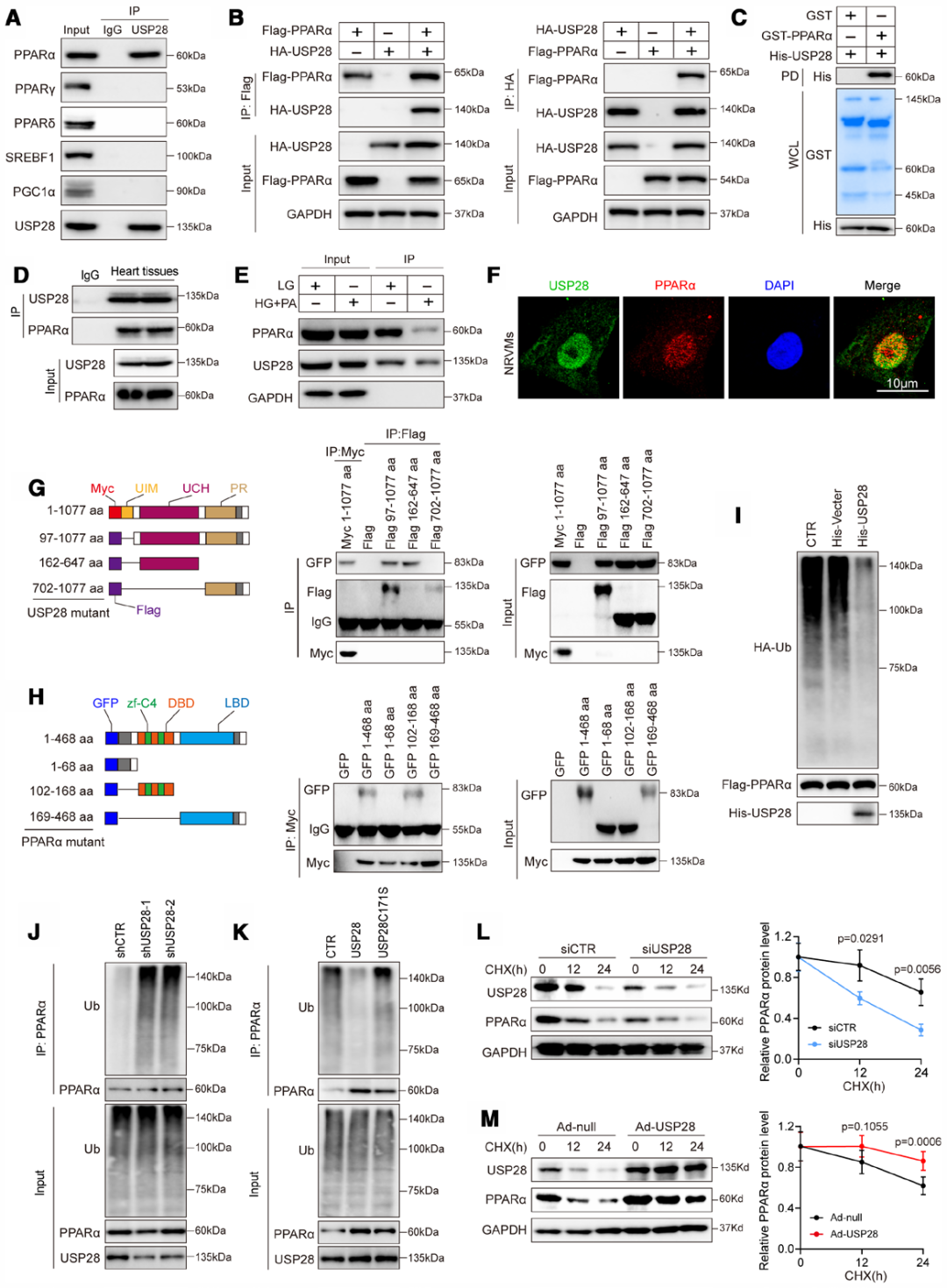
为进一步探究USP28在改善心肌细胞线粒体功能紊乱与脂质代谢异常中所靶向的特异性去泛素化底物,研究人员通过免疫沉淀联合质谱分析,筛选USP28的潜在相互作用蛋白。在转染腺病毒载体Ad-Flag-USP28的HEK293T细胞中,分离USP28相关蛋白复合物并进行胶内质谱检测,共鉴定出226个候选相互作用蛋白,其中PPARα位列候选蛋白列表首位。进一步验证显示,在包括固醇调节元件结合转录因子1(SREBF1)、PPARγ、PPARδ及过氧化物酶体增殖物激活受体γ辅激活因子1α(PGC1α)在内的PPAR家族及脂质代谢相关转录因子中,PPARα是USP28唯一的相互作用蛋白。
研究人员通过免疫共沉淀实验(co-IP)评估USP28与PPARα的直接结合作用。结果显示,在体外共表达外源USP28与PPARα(Flag-PPARα质粒、HA-USP28质粒)的HEK293T细胞中,二者存在特异性相互作用;此外,利用纯化蛋白进行的体外GST pull-down实验进一步证实,USP28可与PPARα直接结合。后续实验在人心脏组织中同样验证了USP28与PPARα的相互作用,且与对照组比较,HG+PA处理的NRVMs中,二者的结合水平显著降低。免疫荧光染色结果显示,在NRVMs中,USP28与PPARα主要共定位于细胞核内。
为明确USP28与PPARα相互作用的分子结构域,研究人员构建了USP28与PPARα的截短突变体。pull-down实验结果表明,USP28的泛素羧基末端水解酶结构域(UCH结构域)与PPARα的DNA结合结构域(DBD结构域)发生了强相互作用。作为一种去泛素化酶,USP28可能通过翻译后修饰调控PPARα的功能。为探究USP28是否可通过去泛素化作用稳定PPARα,研究人员通过去泛素化实验检测PPARα的多泛素化水平。结果显示,体外实验中USP28显著促进了PPARα的去泛素化;在USP28敲低的细胞中,PPARα的泛素化水平显著上调;而在HEK293T细胞中,过表达野生型USP28促进了PPARα去泛素化,酶活性缺失的USP28突变体则无此作用。为明确PPARα上参与USP28介导去泛素化修饰的关键赖氨酸位点,研究人员构建了PPARαDBD内所有赖氨酸位点的突变体,并通过实验证实,赖氨酸152(Lys-152)可能是USP28介导PPARα去泛素化的关键位点。
最后,研究人员利用蛋白质翻译抑制剂环己亚胺(CHX)处理NRVMs,检测PPARα蛋白的半衰期。结果显示,与对照组相比,USP28沉默显著缩短PPARα蛋白的半衰期;而通过腺病毒(Ad-USP28)过表达USP28则延长了PPARα蛋白的半衰期。综合来看,这些结果表明,USP28通过与PPARα特异性相互作用,调控其去泛素化修饰并增强其蛋白稳定性。
图7.USP28可特异性与PPARα相互作用,从而调节PPARα的去泛素化修饰和稳定性
5.USP28通过促进心肌细胞中PPARα介导的Mfn2转录,调控线粒体稳态
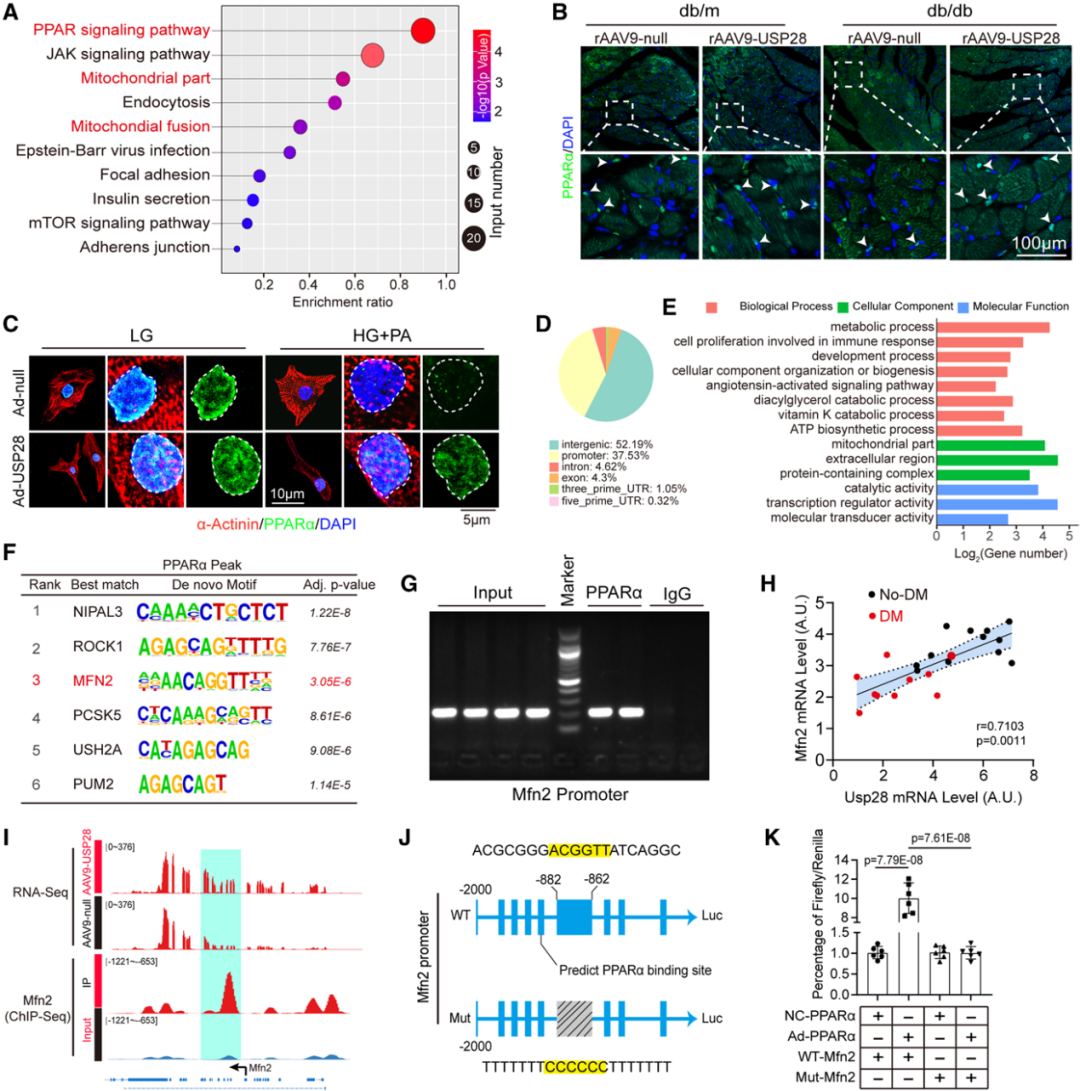
与免疫沉淀联合质谱分析的结果一致,通过KEGG通路对比分析及基因集富集分析(GSEA)发现,DEGs在线粒体代谢通路中显著富集,其中以PPAR信号通路最为突出。为验证上述结果,研究人员在体内实验中检测了PPARs家族蛋白的表达水平:糖尿病心脏中PPARα、PPARδ及PPARγ的表达均降低,而USP28过表达仅能恢复PPARα的表达。此外,USP28还恢复了糖尿病心脏中Mfn1及Opa1的蛋白表达。作者通过心脏组织免疫荧光染色检测PPARα的亚细胞定位发现,糖尿病心脏中PPARα阳性细胞核数量减少;而USP28过表达恢复了糖尿病心脏中PPARα在细胞核内的定位。进一步分离细胞核蛋白检测PPARα核定位水平,结果显示USP28稳定恢复了糖尿病心脏中核内PPARα的表达量。体外实验结果与体内一致:免疫荧光显示,在HG+PA处理的NRVMs中,USP28促进了PPARα向细胞核内转移;同时,USP28还恢复了NRVMs细胞核内PPARα的蛋白水平。
已知PPARα是一种调控基因转录的核转录因子。为探究糖尿病刺激下PPARα的潜在下游靶基因,研究人员在hiPSC-CMs中开展了靶向PPARα的染色质免疫沉淀测序(ChIP-seq)。结果显示,PPARα结合位点分布为:基因的启动子区(37.53%)、内含子区(4.62%)、基因间区(52.19%)及外显子区(4.3%);与PPARα结合峰相关的基因在线粒体结构、ATP合成过程及脂质代谢通路中显著富集。通过对PPARα结合峰的从头基序(de novo motif)分析发现,PPARα的共有基序与Mfn2的基序显著富集。由于Mfn2在线粒体中高表达且在线粒体融合中起关键作用,因此被确定为PPARα的候选下游靶基因。在hiPSC-CMs中通过ChIP进一步验证了PPARα与Mfn2启动子区的直接结合。
在人糖尿病心脏组织中,USP28的mRNA水平与Mfn2的mRNA水平呈正相关,提示Mfn2在USP28介导的心脏保护作用中具有重要意义。此外,为明确PPARα与Mfn2的结合基序,研究人员对Mfn2基因序列中PPARα转录因子的候选结合位点及Mfn2启动子区域进行分析,发现大部分潜在结合位点集中在Mfn2启动子的-882至-862 bp区域。随后,研究人员开展荧光素酶报告基因实验,将野生型或突变型(PPARα结合位点缺失)Mfn2启动子质粒(WT-Mfn2、Mut-Mfn2)转染至PPARα过表达的HEK293细胞后发现,携带WT-Mfn2启动子的载体显著上调了荧光素酶表达,而携带Mut-Mfn2启动子的载体则无此效应。概括而言,这些结果表明:USP28通过促进PPARα向细胞核内定位,进而推动心肌细胞中Mfn2的转录,最终实现对线粒体稳态的调控。
图8.心肌细胞中,USP28通过促进PPARα介导的Mfn2转录来调节线粒体稳态
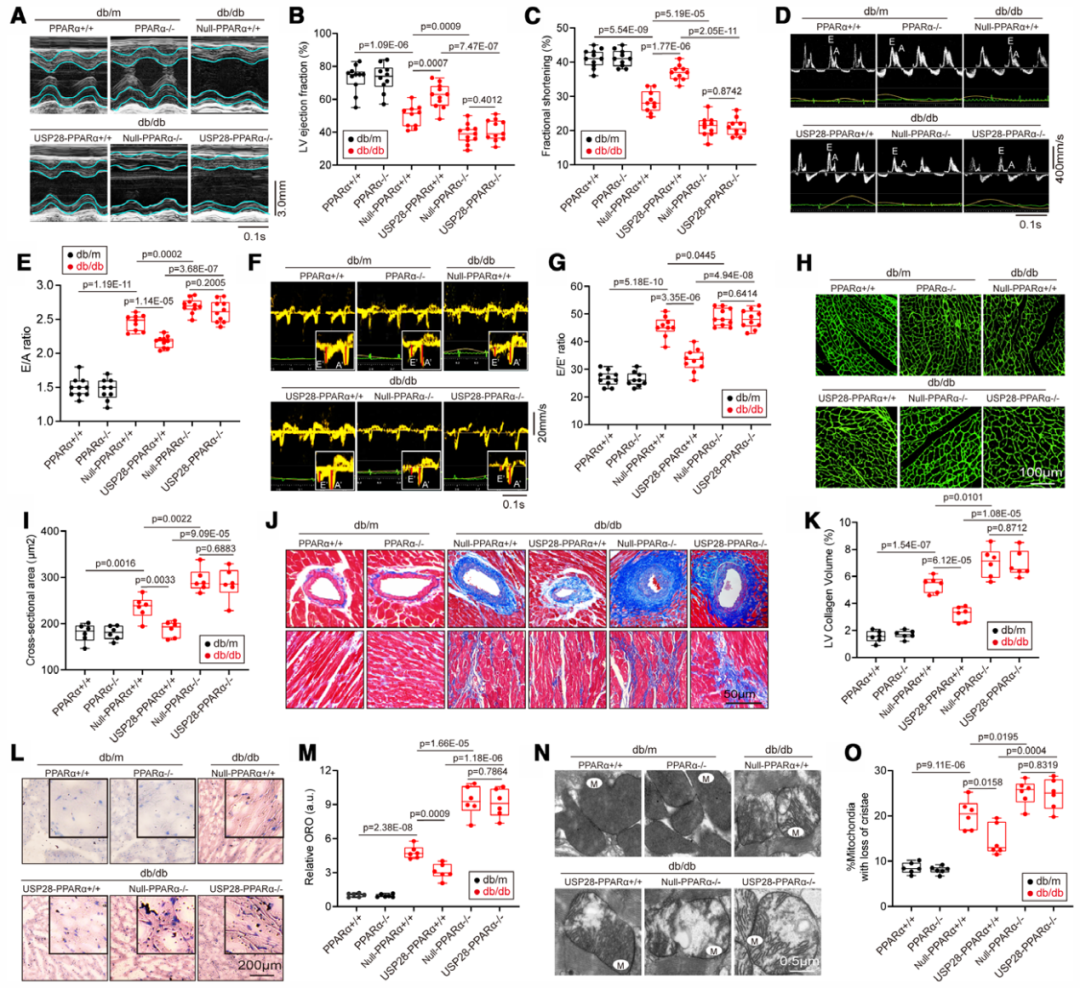
6.PPARα缺失在很大程度上消除db/db小鼠中USP28诱导的心脏保护作用
接下来,研究人员探究了在USP28对抗心脏结构重塑、脂质蓄积及线粒体紊乱的心脏保护作用中,PPARα是否为必需因子。首先,通过将db/m小鼠与PPARα基因敲除小鼠杂交,构建出PPARα基因敲除db/db小鼠(PPARα–/–db/db小鼠)。为研究USP28过表达能否在PPARα缺陷型db/db小鼠中发挥心脏保护作用,向PPARα–/–db/db小鼠体内递送了rAAV9-cTnT-USP28。正如预期的那样,与db/m小鼠相比,db/db小鼠体重显著增加,FBG水平更高。但在db/db或db/m小鼠中,PPARα敲除或USP28过表达对体重和FBG均无影响。经超声心动图检查显示,与db/m小鼠相比,db/db小鼠存在明显的收缩和舒张功能障碍,具体表现为LVEF和LVFS显著降低,以及E/A比值和E/E'比值升高。USP28过表达改善了db/db小鼠的心功能障碍。但研究者发现,在心肌细胞中恢复USP28表达,无法改善PPARα–/–db/db小鼠的收缩和舒张功能障碍。这些结果表明,PPARα缺失消除了USP28对db/db小鼠心功能的改善作用。
在心脏形态方面,同样地,USP28过表达减轻了db/db小鼠的心肌肥厚,具体表现为心脏重量/胫骨长度比值、左心室质量降低,以及心肌细胞横截面积减小。相反,PPARα缺失消除了USP28过表达带来的心脏保护作用。此外,Masson染色结果显示,注射rAAV9-USP28显著减轻了db/db小鼠的心脏纤维化,左心室胶原沉积减少便是有力证据。但在PPARα–/–db/db小鼠中,USP28无法改善心肌纤维化。另外,通过蛋白质印迹检测了db/db小鼠心脏组织中与心肌肥厚相关的蛋白(BNP、β-MHC)以及与纤维化相关的蛋白(CollagenⅠ)表达水平。结果显示,在PPARα–/–db/db小鼠的心肌组织中,BNP、β-MHC和CollagenⅠ的水平显著升高,而USP28过表达无法降低这些蛋白的水平。
已有研究表明,减少细胞内脂滴蓄积可减轻心脏脂毒性表型并改善心功能。本研究中,油红O染色结果显示,USP28过表达减少了db/db小鼠心肌中的脂滴数量。然而,PPARα–/–db/db小鼠的心肌脂滴蓄积更为明显,且USP28无法抑制这种蓄积。
透射电镜结果显示,USP28过表达减少了db/db小鼠心肌中受损线粒体的数量(以线粒体嵴丢失为判断依据)。但PPARα缺失导致受损线粒体数量增加,且这种增加不受USP28过表达的影响。与油红O染色和电镜观察结果一致,脂质组学分析结果也显示,USP28的恢复抑制了糖尿病小鼠心脏的脂毒性,而这种抑制作用依赖于PPARα的激活。正如预期,即使在USP28过表达的2型糖尿病小鼠中,PPARα缺失仍导致FA氧化相关基因的表达降低。
此外,研究人员检测了小鼠心肌组织中的ATP含量。结果显示,PPARα缺失消除了USP28过表达诱导的db/db小鼠心肌ATP生成的增加。同样,USP28表达升高减轻了ROS蓄积,但在体内实验中,PPARα缺失消除了USP28的这一作用。另外,USP28的恢复改善了血管生成,而这种改善作用在PPARα突变小鼠中消失。上述结果表明,PPARα缺失在很大程度上消除了db/db小鼠中USP28诱导的心脏保护作用。
为进一步探究体外实验中USP28的保护作用是否依赖于PPARα,作者从8周龄PPARα突变小鼠(PPARα–/–)和对照小鼠(PPARα+/+)体内提取成年小鼠心肌细胞(AMCMs),并使用α-肌动蛋白(α-actinin)抗体通过细胞免疫荧光法对其进行鉴定。在持续培养基中,用HG+PA处理AMCMs 24小时,以模拟体内高血糖和高血脂环境。经HG+PA处理后,AMCMs中的线粒体肿胀,形态由管状变为肿胀状态;而Ad-USP28通过恢复线粒体平均长度改善了线粒体形态。需特别指出的是,在PPARα–/–AMCMs中,未观察到线粒体形态的改善。随后,研究人员检测了线粒体动力学相关蛋白的水平。蛋白质印迹结果显示,在HG+PA处理的PPARα+/+AMCMs中,USP28过表达升高了Mfn1的水平,并降低Drp1的磷酸化水平。而在HG+PA处理的PPARα–/–AMCMs中,Drp1磷酸化水平显著升高,Mfn1水平降低,且USP28过表达无法改善这些变化。
接下来,作者探究了HG+PA条件下AMCMs的线粒体功能。与预期一致,HG+PA刺激显著降低了PPARα+/+小鼠AMCMs的最大耗氧率(OCR),而USP28过表达部分恢复了OCR。然而,在HG+PA条件下,心肌细胞中USP28过表达无法恢复PPARα–/–小鼠AMCMs的OCR,这提示USP28以PPARα依赖的方式改善应激诱导的线粒体呼吸功能缺陷。综上所述,这些结果表明,USP28减轻线粒体形态和功能缺陷依赖于PPARα。
图9.db/db小鼠中,PPARα的敲除显著削弱了USP28诱导的心脏保护作用
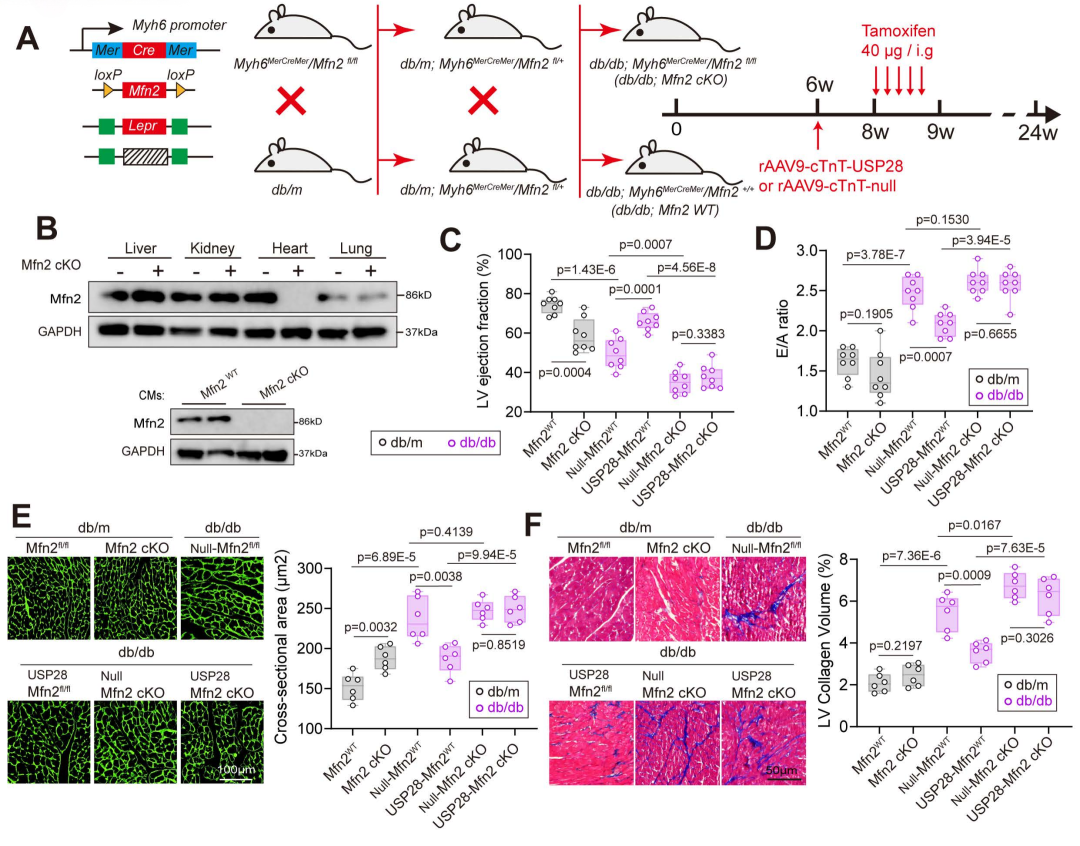
7.心肌细胞中Mfn2功能缺失消除db/db小鼠中USP28恢复表达诱导的心脏保护作用
由于PPARα是关键的代谢基因调控因子,其活性可能影响多条心脏保护通路,因此接下来作者构建了Mfn2功能缺失模型,以验证在糖尿病心脏中,Mfn2是否为USP28介导心脏保护作用所必需。通过将db/m小鼠与Myh6MerCreMer/Mfn2fl/fl小鼠杂交,构建出可诱导的心肌特异性Mfn2缺陷型db/db小鼠(db/db-Myh6MerCreMer/Mfn2fl/fl)。此外,通过蛋白质印迹检测心脏匀浆和分离的心肌细胞,证实他莫昔芬(tamoxifen)注射可成功诱导Mfn2缺失。
在Mfn2fl/fl db/db小鼠中,注射rAAV9-USP28提高了LVEF并改善E/A比值;但在Mfn2功能缺失的db/db小鼠中,USP28无法改善其收缩和舒张功能障碍。随后发现,注射rAAV9-USP28显著减轻了db/db小鼠的心肌肥厚和纤维化,心肌细胞横截面积减小、左心室胶原含量降低便是直接证据。然而,在Mfn2突变型db/db小鼠中,USP28无法改善心肌肥厚和纤维化。他莫昔芬诱导心肌细胞中Mfn2功能缺失导致收缩功能障碍和心肌肥厚,但对E/A比值和左心室胶原沉积无影响。在线粒体缺陷和脂质代谢方面,Mfn2缺失后,USP28过表达无法减轻心脏线粒体损伤和脂质蓄积。总而言之,这些数据表明,心肌细胞中Mfn2诱导性缺失消除了db/db小鼠中USP28恢复表达诱导的心脏保护作用。
图10.USP28以Mfn2依赖的方式改善糖尿病性心力衰竭和心脏重构
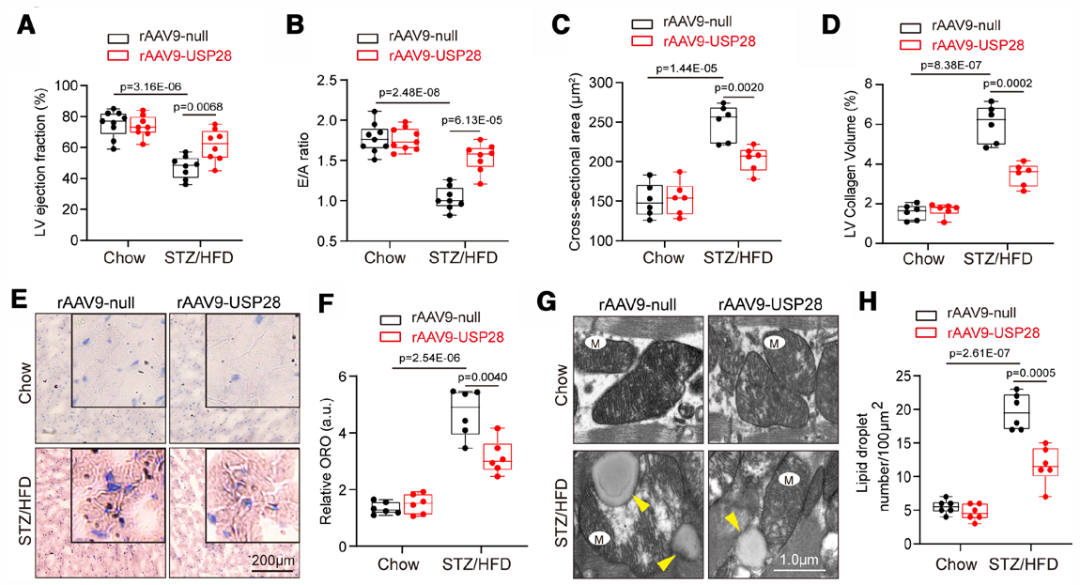
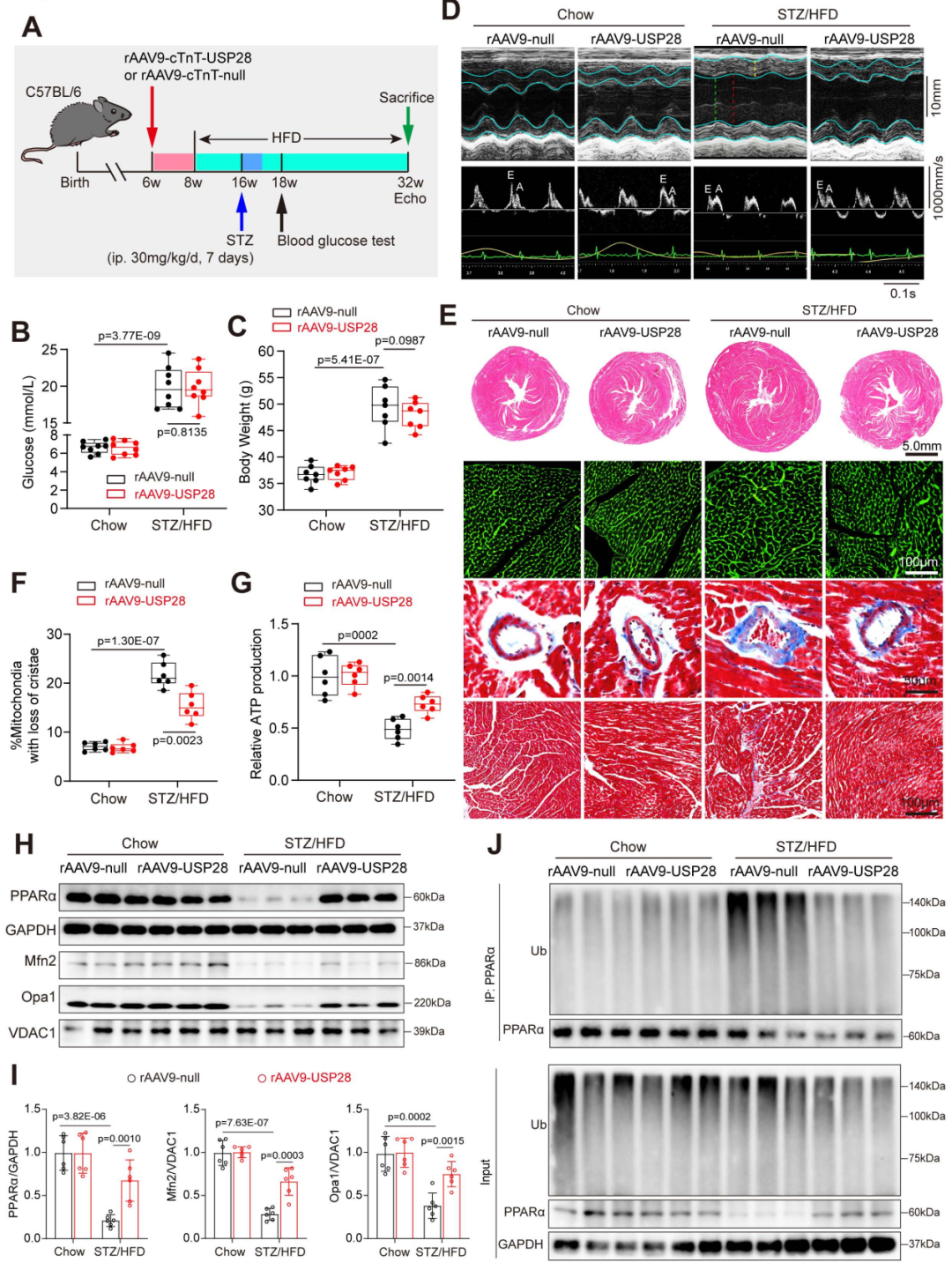
8.恢复心肌USP28表达可改善高脂饮食联合链脲佐菌素诱导的2型糖尿病小鼠糖尿病性心力衰竭及线粒体紊乱
为进一步评估恢复USP28表达对DCM的作用,研究人员采用高脂饮食(HFD)联合链脲佐菌素(STZ)构建了另一种2型糖尿病小鼠模型(HFD喂养24周;HFD喂养8周后连续7天腹腔注射STZ)。在给予STZ/HFD处理前,向6周龄C57BL/6J雄性小鼠体内递送rAAV9-cTnT-USP28,以诱导心肌细胞中USP28过表达。正如预期的那样,与正常饮食小鼠(喂养标准基础饲料,Chow)相比,STZ/HFD小鼠体重显著增加,且FBG更高。与预期一致,在STZ/HFD小鼠和Chow小鼠中,USP28过表达对体重和FBG均无影响。超声心动图分析显示,STZ/HFD小鼠的LVEF和E/A比值明显降低;而USP28过表达恢复了左心室收缩和舒张功能,不过在Chow小鼠中,USP28过表达对心功能无显著影响。这些结果表明,USP28过表达显著减轻了糖尿病心脏的收缩和舒张功能障碍。
接下来,研究人员探究了USP28过表达对DCM中心脏重塑的影响。组织学分析显示,USP28过表达改善了STZ/HFD小鼠的左心室肥厚,减小心肌细胞体积,并减轻心肌纤维化。这些结果进一步表明,USP28过表达减轻了2型糖尿病小鼠的病理性心脏重塑。
为研究USP28过表达对HFD/STZ诱导的心脏脂质代谢异常的作用,研究人员通过油红O染色评估糖尿病心脏中的脂质含量。结果显示,STZ/HFD小鼠心肌中的脂质蓄积增加,而USP28过表达显著抑制了这一变化。此外,透射电镜结果显示,USP28过表达显著减少STZ/HFD小鼠心肌中的脂滴数量,并降低嵴丢失线粒体的比例。这些结果提示,USP28过表达改善了2型糖尿病小鼠心肌脂质利用障碍。不仅如此,USP28还恢复了2型糖尿病小鼠心肌的ATP生成。
随后,研究者检测了小鼠心脏中PPARα及线粒体融合相关关键蛋白的表达水平。蛋白质印迹结果显示,在STZ/HFD小鼠中,USP28过表达上调了Mfn2、Opa1和PPARα的表达水平,而在Chow小鼠中无显著影响。同时,通过内源性免疫沉淀实验,发现PPARα表达水平的升高与其泛素化降解减少有关。不出所料,USP28过表达对Chow小鼠的PPARα泛素化水平无显著影响,但显著抑制了STZ/HFD诱导的2型糖尿病小鼠心脏中PPARα的泛素化水平。由此看来,这些结果表明,恢复心肌USP28表达可改善HFD/STZ诱导的2型糖尿病小鼠的糖尿病性心力衰竭及线粒体形态和功能缺陷。
图11.HFD/STZ诱导的2型糖尿病小鼠中,心脏USP28的恢复可抵抗糖尿病性心力衰竭和线粒体紊乱
图12.心脏USP28的恢复可对抗HFD/STZ诱导的2型糖尿病小鼠的糖尿病性心力衰竭
总结
本研究通过db/db小鼠、STZ/HFD小鼠两种独立啮齿动物模型及体外细胞实验,证实USP28可作为糖尿病心脏的潜在调控因子,为线粒体稳态在DCM结构及代谢重塑调控中的作用提供了实验证据。研究结果表明,糖尿病心脏中心肌细胞USP28表达下调会扰乱脂质代谢与线粒体功能,同时还会抑制PPARα-Mfn2介导的线粒体融合信号通路(PPARα通过转录调控Mfn2表达,进而促进线粒体融合)。然而,通过心肌特异性AAV载体恢复USP28表达后,可促进PPARα的去泛素化修饰以维持其蛋白稳定性,进而启动Mfn2的转录过程并恢复线粒体稳态,最终改善DCM的心脏重塑与功能障碍。因此,本研究揭示了DCM发病机制的新见解,并通过证实USP28在维持线粒体功能及糖尿病心脏功能中的关键作用,为DCM提供了一种具有潜力的治疗策略。
图13.模式图

通讯作者介绍
唐其柱,武汉大学人民医院心血管内科,医学博士,教授(二级)、主任医师(一级),博士生导师。心血管病研究所副所长、代谢与相关慢病湖北省重点实验室主任。中央“万人计划”百千万工程领军人才,卫生部有突出贡献中青年专家,湖北省首批医学领军人才。中国医师协会心力衰竭专业委员会副主任委员、湖北省医学会心血管病分会常委、湖北省心脑血管病防治办公室主任。擅长于心血管内科疾病的诊治,特别是在急慢性心力衰竭以及各种心肌病的诊治方面具有较高的造诣和经验。在高血压、冠心病等重大慢性病的预防和管控方面有深入的研究。主持研究的有关心力衰竭防治两项成果获湖北省科技进步一等奖,参与研究的3项有关心律失常防治的成果获国家科技进步二等奖。发表论文260余篇(SCI收录120余篇),主编专著6部、参编专著15部。先后主持国家自然科学基金重点项目、国家重点研发计划项目等10余项。


了解产品及服务
请扫码添加客服微信:BrainVTA2020

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK