2025-06-16 阅读量:3118
编者按
随着阿尔茨海默病(AD)研究的不断深入,神经免疫相互作用在AD发病机制中的关键作用愈发受到关注。从外周免疫细胞的浸润到中枢神经系统内的炎症反应,从关键免疫介质的失调到血脑屏障(BBB)的破坏,这些复杂的神经免疫过程交织在一起,共同推动了AD的进展。然而,目前对于这些神经免疫机制的理解仍存在诸多空白,尤其是在不同阶段外周免疫细胞的具体作用以及如何精准调控这些免疫过程以实现治疗目的方面。
因此,小编在这里给大家推荐这篇厦门大学医学院神经科学研究所王鑫教授团队于2025年2月发表在Molecular Neurodegeneration上题为“Peripheral and central neuroimmune mechanisms in Alzheimer’s disease pathogenesis”的综述,该文系统梳理了神经系统与免疫系统在AD中的双向通讯,详细阐述了BBB破坏、外周免疫细胞浸润、关键免疫介质失调等多方面内容,深入探讨了脑膜淋巴管功能、免疫衰老对T细胞启动的影响、CD8⁺ T细胞的复杂作用等前沿问题,并对靶向神经免疫相互作用的新治疗策略进行了展望。

随着全球人口老龄化加剧,AD这一主要神经退行性疾病的患病率持续攀升,已成为严峻的公共卫生挑战。既往研究多聚焦于淀粉样斑块和神经原纤维缠结,而近期研究发现,外周免疫系统在AD发病机制中扮演关键角色,促使人们重新审视该疾病的潜在机制。BBB的完整性对AD进程影响重大,一旦受损,外周免疫细胞便会侵入中枢神经系统(CNS),加剧神经炎症,加速认知功能衰退。新兴研究揭示,外周免疫细胞与中枢神经系统驻留小胶质细胞之间存在复杂双向通讯,T细胞、巨噬细胞、自然杀伤细胞、中性粒细胞及B细胞等多种免疫细胞可调节AD大脑的神经炎症微环境,形成自我持续的炎症级联反应。此外,促炎细胞因子、载脂蛋白E4(APOE4)、β2微球蛋白(B2M)及小胶质细胞受体TREM2等关键免疫介质的失调,进一步扰乱神经免疫平衡,影响AD进展。本综述系统梳理了外周免疫系统在AD中作用机制的新研究进展,重点阐述免疫-中枢神经系统通讯机制及潜在治疗靶点,旨在明确未来研究方向,探索针对AD神经免疫相互作用的创新治疗策略。
一、外周免疫细胞在AD中的作用及机制
1.CD8⁺ T细胞
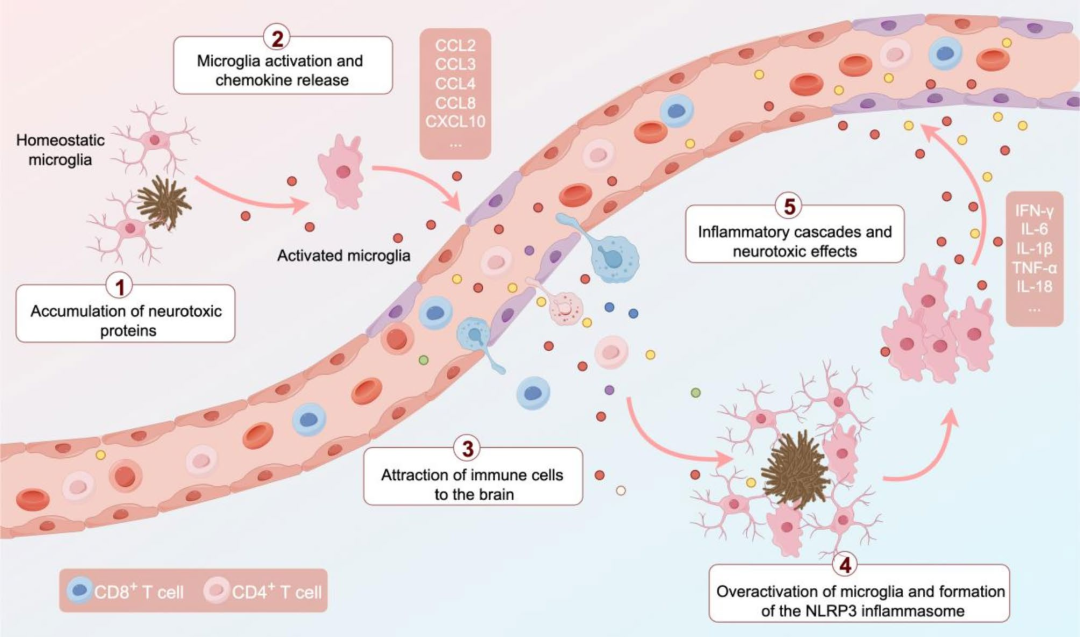
CD8⁺ T细胞的脑内浸润
BBB由脑微血管内皮细胞的紧密连接构成,是一种高度选择性结构,通过限制外部物质和免疫细胞进入CNS,从而维持内环境稳态。其独特的细胞组成(由特化内皮细胞构成,周细胞和星形胶质细胞终足提供支持)和有限的通透性,确保仅特定分子能够穿过。实验模型研究表明,在AD病理状态下,BBB的保护功能会逐渐受损,但这一发现仍需在人体样本中进一步验证。随着疾病进展,β-淀粉样蛋白(Aβ)不仅在脑实质中形成斑块,还会在脑膜和脑血管中沉积,引发脑淀粉样血管病(CAA),进一步破坏BBB,导致血管功能障碍、缺血、微出血及外周免疫细胞浸润增加。
在AD患者中,BBB功能失调促使外周CD8⁺ T细胞侵入CNS,从而加剧神经炎症和疾病进展。尽管其具体入脑机制尚未完全明确,但研究已发现硬脑膜淋巴管、脉络丛和脑血管等潜在路径。研究显示,AD患者血液中CD8⁺ T效应记忆CD45RA(TEMRA)细胞数量增加,且与认知功能呈负相关。此外,脑脊液中还检测到携带Epstein-Barr病毒(EBV)特异性T细胞受体(TCR)的克隆扩增CD8⁺ TEMRA细胞,这提示EBV感染可能增加AD风险。动物模型(如Tau转基因小鼠)中也观察到CD8⁺和CD4⁺ T细胞的克隆扩增现象。针对早期AD,调控CD8⁺ T细胞迁移和功能对遏制BBB恶化及疾病进展至关重要。3D人神经免疫轴模型显示,抗CXCR3(趋化因子受体3)中和抗体阻断CXCL10(趋化因子配体10)-CXCR3相互作用,可显著减轻CD8⁺ T细胞浸润引发的AD病理,但3D培养系统难以完全模拟体内免疫系统的复杂性,因此需结合动物模型实验以深化对CD8⁺ T细胞机制的理解。此外,研究还表明,联合使用CCL2(单核细胞趋化蛋白-1)和CCL8(单核细胞趋化蛋白-2)的中和抗体可抑制免疫细胞穿越BBB,而阻断CCL3(巨噬细胞炎症蛋白-1α)能部分减少老年脑中CD8⁺ T细胞募集。因此,通过调控和抑制CD8⁺ T细胞的浸润能力及功能活性,有望延缓AD的病理进程。
CD8⁺ T细胞与小胶质细胞的相互作用
在AD大脑中,小胶质细胞对Aβ、tau(微管相关蛋白)等神经毒性蛋白沉积高度敏感。激活后的小胶质细胞释放趋化因子,吸引CD8⁺ T细胞浸润脑组织;而CD8⁺ T细胞分泌穿孔素、颗粒酶等细胞毒性因子,进一步激活小胶质细胞,促使其产生干扰素-γ(IFN-γ)、白细胞介素-1β(IL-1β)和肿瘤坏死因子-α(TNF-α)等促炎细胞因子。这些细胞因子不仅加剧炎症反应,还会促进Aβ生成与聚集、抑制其清除,进而加剧BBB功能障碍,并促进CD8⁺ T细胞进一步渗入脑组织,形成自我加剧的恶性循环,加速神经退行性进展。在tau蛋白病小鼠模型和AD患者大脑中,tau病理富集区域内的CD8⁺ T细胞显著增多。小胶质细胞介导的T细胞渗入导致神经元功能障碍和死亡,破坏神经通信和记忆形成。在衰老大脑中,由CD8⁺ T细胞引发的炎症级联反应是神经退行性过程的关键驱动因素。CD8⁺细胞毒性T细胞是IFN-γ的主要来源,其通过诱导IFN响应性小胶质细胞触发中枢神经系统的神经炎症事件。IFN-γ还直接影响少突胶质细胞、小胶质细胞和神经干细胞,表明CD8⁺ T细胞可能通过促进少突胶质细胞死亡和轴突退化,导致认知障碍和运动功能下降。在5×FAD小鼠模型中,CD8⁺ T细胞与Aβ斑块附近的小胶质细胞紧密关联,且外周B细胞参与其募集过程。因此,靶向干预小胶质细胞与CD8⁺ T细胞的相互作用,如小胶质细胞或T细胞耗竭、抑制IFN-γ信号通路,或可缓解tau介导的神经退行性病变,减轻AD相关神经炎症与神经退行性改变。
图1. T细胞与小胶质细胞相互作用介导的神经炎症自我持续反馈环
2.CD4⁺ T细胞
CD4⁺ T细胞具有显著可塑性,能根据细胞因子环境分化为不同效应亚群,在AD发病机制中发挥不同作用。其中,1型和17型辅助性T细胞(Th1和Th17)等促炎亚群有助于BBB破坏和增强小胶质细胞活化,加剧AD相关的神经病理;而Th2细胞和调节性T细胞(Tregs)等抗炎亚群则具备神经保护功能,能减轻神经炎症。CD4⁺ T细胞应答的这种功能二分法,不仅揭示了AD免疫调控网络的复杂性,也为AD免疫干预提供了潜在靶点。
CD4⁺ T细胞的促炎作用
在AD研究中,Th1细胞在APP/PS1小鼠(转基因AD模型小鼠)中可通过分泌IFN-γ激活小胶质细胞,促进淀粉样斑块清除,但过度激活的Th1细胞会导致IFN-γ和TNF-α过量分泌,引发Aβ积累与小胶质细胞过度活化,加重神经炎症与认知损伤,且过继转移Aβ刺激的Th1细胞会增加脑内Aβ负担、损害突触可塑性。此外,抗Aβ单克隆抗体和Aβ疫苗的疗效有限,可能与其诱导Th1细胞介导的炎症反应有关,这种反应可能导致过度的免疫激活。
Th17表现出强大的脑浸润能力,并分泌IL-17A、IL-23、IL-21、IL-6、IFN-γ等多种细胞因子,引发小胶质细胞过度激活与其它免疫细胞募集为特征的神经炎症反应。过继转移Aβ特异性的Th1和Th17细胞(能够通过特异性T细胞受体识别Aβ)会加速小鼠记忆障碍、并提高血液中TNF-α、IFN-γ和IL-17的水平。另外,IL-17还可通过增强神经炎症、抑制小胶质细胞吞噬作用、加剧淀粉样蛋白沉积推动AD进展,而中和IL-17能缓解认知与突触功能障碍。
Th22细胞能够分泌IL-22而不共表达IL-17或IFN-γ,升高的IL-22水平会激活AD脑内胶质细胞,促使促炎因子产生及淋巴细胞浸润到脑实质;Th9细胞在AD中显著上调,其分泌的IL-9与TGF-β1(转化生长因子-β1)协同促进初始CD4⁺ T细胞向Th17细胞分化,而Th17细胞在AD患者外周血单个核细胞中也有所增加。
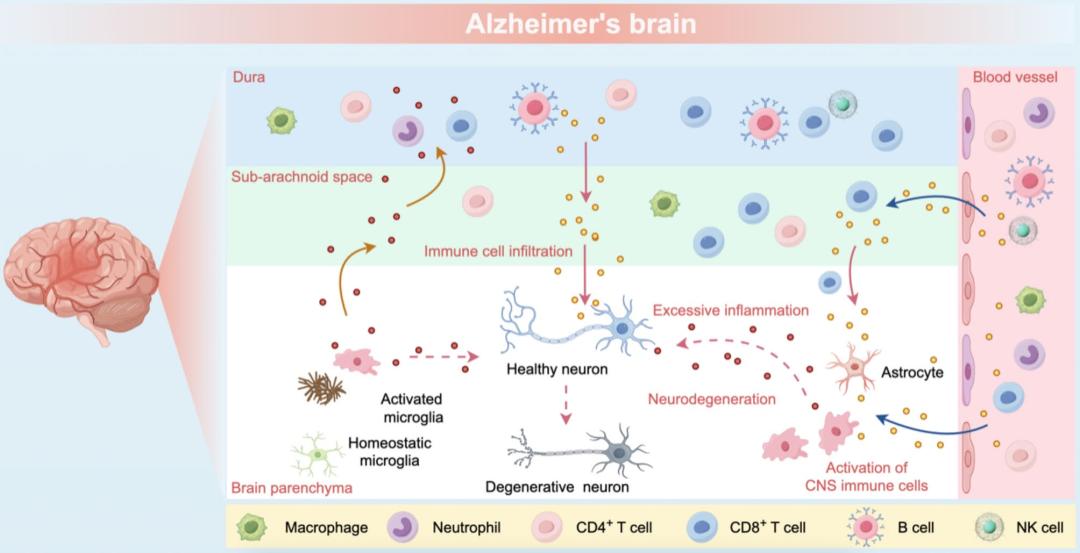
图2. 外周免疫对AD患者中枢神经系统的影响
CD4⁺ T细胞的抗炎作用
在AD中,Aβ刺激的Th2细胞能够抑制Th1和Th17细胞产生IFN-γ,并下调小胶质细胞中CD86和CD40等共刺激分子的表达,从而减轻促炎反应。在APP/PS1小鼠模型中,注入纯化的Th2细胞可以降低血浆中IFN-γ、TNF-α、GM-CSF(粒细胞-巨噬细胞集落刺激因子)、IL-2、IL-4和Aβ的水平,同时改善认知功能并缓解病理特征。
Tregs通过分泌IL-10、TGF-β等抗炎细胞因子,抑制效应T细胞活化和增殖,在免疫调控中起关键作用。人类AD脑尸检显示Treg数量较非AD对照减少,伴随促炎细胞因子(如TNF-α、IL-1β和IL-6)的过量产生,导致神经炎症效应增强;APP/PS1小鼠实验表明,过继转移Aβ特异性Th1和Th17细胞会削弱CNS和外周血中Tregs数量与功能,加剧神经炎症。
在APP/PS1小鼠中,低剂量人IL-2的外周给药可选择性促进Tregs扩增,恢复Treg/Th17平衡,增加与淀粉样斑块相关的小胶质细胞,减少淀粉样斑块和神经炎症,从而增强认知功能,相关疗法已进入AD治疗的2期临床试验(NCT06096090)。另一种潜在的治疗策略是直接转移Tregs:Aβ刺激后Tregs的自主体外扩增和转移,能在5×FAD小鼠中降低Aβ负荷、抑制小胶质细胞过度激活,同时下调促炎细胞因子和补体级联反应,从而改善认知障碍、Aβ积累、tau过度磷酸化和神经炎症。但也有研究显示,短暂耗竭FOXP3⁺ Tregs(FOXP3是Tregs的特异性标志物)可触发IFN-γ依赖的全身免疫反应并激活脉络丛,促进单核细胞衍生巨噬细胞(MDMs)和Tregs渗入淀粉样病变的脆弱脑区域,同样能减轻Aβ负担并改善5×FAD小鼠的认知功能。这些发现提示,Treg调控在AD中具有复杂双面性,需结合具体病理背景和免疫环境制定精准治疗策略,目前通过TGF-β和IL-10信号通路增强Treg抑制功能的分子靶向方法仍在探索中。
3.其他外周免疫细胞
巨噬细胞:包括边界相关巨噬细胞(BAMs)和MDMs,在AD发病机制中发挥关键作用。BAMs位于大脑边缘区域,通过促进淋巴系统清除,对维持大脑稳态至关重要。但随年龄增长,其主要组织相容性复合体II类分子(MHC-II)表达增强、细胞外基质降解能力受损,导致血管和类淋巴系统功能障碍,增加AD风险,且在具有淀粉样病变的AD小鼠模型中,BAMs缺失会加剧Aβ沉积与小胶质细胞活;MDMs随年龄增长而积累并参与Aβ清除,但其Aβ摄取能力会随着年龄增长和AD进展降低,部分是由于吞噬受体(如TREM2)表达减少。TREM2基因变异与AD风险相关,TREM2⁺巨噬细胞聚集于Aβ斑块,TREM2缺陷小鼠炎症与病理减轻。在TREM2缺陷的AD小鼠模型中,CD45hiLy6C⁺(高表达白细胞共同抗原,淋巴细胞抗原6C阳性)巨噬细胞数量大幅减少,炎症减轻,淀粉样和tau病理改善。通过抗PD-1(程序性死亡受体-1)抗体阻断免疫检查点,可招募MDMs进入大脑,促进淀粉样斑块的清除,恢复5×FAD小鼠的认知功能。值得注意的是,PD-1/PD-L1(程序性死亡配体-1)阻断不仅影响MDMs,还破坏产生IFNγ的T细胞与其靶细胞之间的负信号,广泛增强免疫反应。基于此,人源化IgG1抗体IBC-Ab002(抑制相关免疫检查点蛋白PD-L1)已进入1期临床试验(NCT05551741)。人类海马尸检组织的转录组和免疫组化分析显示,AD晚期单核细胞衍生的巨噬细胞浸润脑实质。但在淀粉样病变的AD小鼠模型中,外周单核细胞替代脑中小胶质细胞不影响斑块负荷,这一发现引发了人们对外周单核细胞与小胶质细胞在Aβ清除中功能差异的探讨。
自然杀伤细胞(NK细胞):作为天然免疫系统的关键组成部分,在AD发病机制中发挥复杂且看似矛盾的作用。在3×Tg-AD小鼠模型中耗竭NK细胞具神经保护作用,包括减轻神经炎症、增强神经发生和改善认知功能。相反,在APP/PS1小鼠模型中增强NK细胞功能可减轻脑内Aβ沉积,提高认知表现。近期的临床研究,尤其是ASK-AD试验(NCT04678453),探索了自体NK细胞疗法(SNK01)在AD患者中的治疗潜力。初步结果表明,SNK01治疗可能导致脑内Aβ和tau蛋白水平降低,同时减轻神经炎症。
中性粒细胞:在AD中公认的脑灌注不足病理过程中起关键作用,其通过黏附于毛细血管段,导致脑血流量(CBF)进行性减少甚至阻塞。此外,中性粒细胞通过破坏紧密连接蛋白(如occludin和claudins)破坏BBB完整性,增加BBB通透性。在5×FAD和3×Tg-AD等转基因AD小鼠模型中,中性粒细胞外渗并在淀粉样沉积区域积聚,伴随着中性粒细胞胞外陷阱(NETs)的形成和IL-17的表达。Aβ42肽诱导淋巴细胞功能相关抗原1(LFA-1)的高亲和力构象,促进中性粒细胞迅速黏附于整合素配体,进而加剧AD病理和认知衰退。
针对中性粒细胞的治疗手段已显示出希望。在疾病早期阶段短暂耗竭中性粒细胞可带来持续的认知改善,而给予Ly6G(淋巴细胞抗原6G)抗体或LFA-1抑制剂可增强脑血流,减轻AD样神经病理,改善认知功能。此外,LFA-1缺陷的转基因AD小鼠表现出对认知恶化的抵抗力,并且胶质细胞增生减少。LFA-1不仅对NK细胞反应至关重要,还被其他外周免疫细胞(包括T细胞)表达,在激活、黏附和迁移中发挥重要作用。LFA-1参与这些多样的免疫细胞功能,暗示了其在AD病理中的潜在重要性,值得进一步研究。
B细胞:在AD患者中被激活,在外周和脑实质中积累,产生靶向Aβ的免疫球蛋白。虽然靶向Aβ的B细胞衍生免疫球蛋白可能阻止斑块形成和疾病进展,但它们也可能同时损害小胶质细胞功能,加剧AD病理。B细胞在AD中的作用不仅限于免疫球蛋白的产生,如在老年3×Tg AD小鼠的颈部淋巴结中,活化B细胞数量的增加与疾病进展的相关性证明了这一点。在5×FAD和APP/PS1小鼠中,B细胞耗竭与疾病进展减缓、认知和运动缺陷改善以及淀粉样蛋白负荷减少相关。然而,矛盾的是,在疾病早期阶段进行B细胞耗竭会加速AD小鼠模型的认知衰退和淀粉样蛋白负荷增加,突显了B细胞在AD发病机制中复杂且阶段依赖性的作用。
黏膜相关恒定T(MAIT)细胞:作为一类天然样T细胞亚群,能够通过组织相容性复合体I类分子(MHC-I)相关蛋白MR1识别微生物来源的代谢产物,尤其是维生素B衍生物。在5×FAD小鼠模型中,MAIT细胞数量逐渐增加并表现出激活特征。AD患者和5×FAD小鼠在淀粉样斑块周围的小胶质细胞中均表现出显著升高的MR1表达。值得注意的是,MR1缺陷小鼠的淀粉样斑块形成明显减少。
γδT细胞:作为天然免疫细胞的另一个亚群,在健康脑膜中是IF-17A的主要来源,它们在CA1海马神经元中支持突触可塑性,并促进短期记忆。然而,在3×Tg AD小鼠模型中,γδT细胞在脑实质和脑膜中大量积累,与认知能力下降相关。值得注意的是,这些小鼠的CNS中产生IL-17的γδT细胞显著增加,其积累与早期短期记忆障碍和突触功能障碍相关。这些发现表明,γδT细胞可能加剧神经炎症,并促进早期AD病理进展。
二、AD中免疫因子的失调
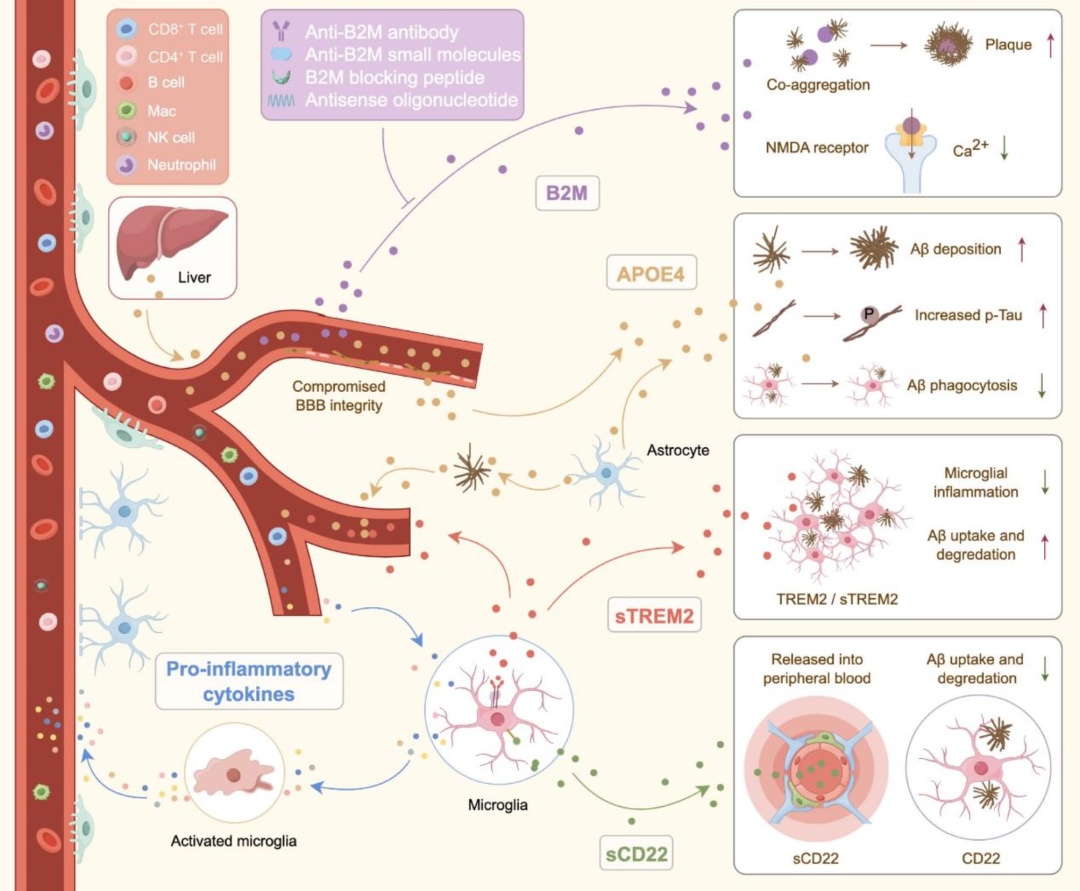
免疫因子失调在AD病理生理中起关键作用,显著影响神经免疫轴。异常的神经免疫调控不仅通过破坏BBB完整性促使外周免疫细胞侵入CNS,还直接加剧神经退行性进程。促炎细胞因子、APOE4、B2M、TREM2和CD22(B细胞受体共受体)等关键神经免疫介质,在调节神经炎症反应及维持CNS与外周免疫稳态交互中至关重要。这些神经免疫调节因子的紊乱会加剧神经炎症,破坏BBB功能,并通过增加外周免疫细胞的浸润来加速神经退行性变。因此,阐明免疫因子失调的机制并开发靶向免疫调节干预手段,对推动延缓和/或阻止神经退行性疾病进展的新型治疗策略至关重要。
图3. AD发病机制中免疫因子的失调
1.促炎细胞因子
IL、TNF、IFN等促炎细胞因子由外周免疫细胞分泌,AD患者血浆中这些因子水平显著升高,促使其通过神经退行性变进程中易渗漏的BBB重新分布。在5×FAD小鼠模型中,I型干扰素(IFN-I)可激活小胶质细胞并诱导突触修剪,通过特异性阻断剂或中和抗体选择性抑制干扰素α/β受体(IFNAR),能够减轻小胶质细胞增生并防止突触丢失。II型干扰素IFN-γ在AD中作用呈双向性:生理水平下可增强小胶质细胞吞噬能力、促进Aβ清除、提高认知功能,还能募集单核细胞衍生巨噬细胞到CNS,清除淀粉样斑块、改善认知;但慢性IFN-γ诱导的炎症会破坏BBB完整性,使外周炎症介质渗入CNS,从而加剧神经炎症、神经元损伤和认知衰退。此外,促炎环境会诱导NLRP3(NOD样受体家族含PYRIN结构域蛋白3)上调并激活,形成NLRP3炎症小体,大量产生促炎细胞因子。在AD中,NLRP3炎症小体主要在先天免疫细胞(如中枢小胶质细胞)中活化,其过度激活引发的炎症级联反应会加重神经元损伤。研究显示,16月龄APP/PS1转基因小鼠中,Nlrp3基因缺失可显著增强小胶质细胞对Aβ的吞噬能力,减少淀粉样斑块并改善空间记忆;在Tau22转基因(AD模型)小鼠中,Nlrp3缺失也能减轻tau蛋白过度磷酸化与聚集,缓解Aβ诱导的tau病理变化。
2.B2M
B2M作为MHC-I复合物的关键组分,循环中主要以非共价结合单体蛋白形式存在并经肾小球滤过清除。病理状态下(如长期透析的慢性肾病),B2M可形成淀粉样纤维引发淀粉样病变及相关疾病。近年研究发现,B2M在AD及唐氏综合征(DS)中通过中枢和外周途径影响认知功能。在AD中,升高的B2M与Aβ共聚集促进淀粉样病变,促进斑块形成和传播,加剧神经毒性和神经炎症,加速神经退行性变及Aβ病理扩散。AD小鼠模型中,通过基因敲除、反义寡核苷酸(ASO)给药或抗体干预降低B2M水平,可显著减轻认知障碍和淀粉样斑块负荷。其跨BBB特性为治疗提供新方向,如外周注射低剂量抗B2M抗体可有效降低脑内B2M并改善认知,避免了高剂量Aβ抗体疗法的常见副作用。
DS患者通常在中年时期发展为AD,提示两者病理机制的重叠。DS中,血浆升高的B2M穿越BBB后抑制N-甲基-D-天冬氨酸(NMDA)受体活性,破坏突触兴奋/抑制平衡,诱发认知功能障碍。机制研究表明,B2M直接与NMDA受体GluN1亚基相互作用削弱其功能,而竞争性抑制该相互作用的治疗性肽段GluN1-P2可显著改善DS和衰老小鼠模型的突触可塑性与认知能力。联体共生和血浆转移实验证实,循环B2M升高是DS患者及模型动物认知损伤的关键介质,靶向清除外周B2M(如抗体中和、联体循环交换)可逆转相关缺陷。
作为系统性衰老因子,B2M在血浆和脑脊液中的水平随年龄增长升高,加剧衰老和神经退行性疾病中的认知衰退。其与Aβ的共聚集增强了神经毒性,这使得淀粉样蛋白级联假说的完善成为必要。而其与NMDA受体的相互作用使其成为潜在治疗靶点,为开发针对B2M介导的小分子抑制剂和抗体疗法提供了机制依据。
3.APOE4
人类19号染色体编码的APOE4基因变异是晚发性AD最重要的遗传风险因素,其112和158位精氨酸残基(Arg112/Arg158)使其区别于APOE3(Cys112(112位半胱氨酸残基)/Arg158)和APOE2(Cys112/Cys158)。APOE4对CNS和外周免疫均有影响,在认知正常的APOE4携带者中,脑组织和脑脊液(CSF)出现血浆蛋白预示BBB完整性在认知下降前已受损。它通过削弱内皮紧密连接、增加BBB通透性、促进神经炎症,减少毛细血管基底膜面积、提升血管壁及血管周围凝血酶水平等方式破坏BBB,还会促进Aβ沉积、加重CAA,并改变Aβ清除途径,延缓Aβ清除,而APOE2和APOE3则利于BBB快速清除Aβ。此外,APOE4会促使脑膜淋巴管过早萎缩,阻碍Aβ清除。
在免疫方面,APOE4加速中性粒细胞免疫衰老,导致产生IL-17的免疫抑制性中性粒细胞浸润女性AD患者脑部,在人APOE4基因敲入APP/PS1小鼠中,敲除中性粒细胞中的APOE4,可逆转其免疫抑制表型并降低了IL-17信号传导,这种改变恢复了小胶质细胞的反应、减轻了淀粉样病变并提升认知功能。动物实验显示,肝脏表达的APOE4会加重脑淀粉样病变,APOE3则相反,提示外周的APOE4损害了脑血管功能,影响了突触可塑性和认知,靶向外周APOE4或可成为AD治疗策略,如血管紧张素受体阻滞剂等靶向疗法有望改善BBB功能、延缓AD进展。
APOE主要由CNS中的星形胶质细胞和活化小胶质细胞合成分泌,在PS19-APOE4小鼠模型(AD小鼠模型)中,不同细胞中敲除APOE4呈现差异化作用:星形胶质细胞特异性敲除可减少疾病相关基因表达、抑制异常突触吞噬;神经元特异性敲除显著减轻tau病理、改善神经退行性变;小胶质细胞特异性敲除则增强APP/PS1小鼠的Aβ清除能力。这些细胞类型特异性的效应揭示了APOE4在AD发病机制中的不同贡献。
4.TREM2
TREM2作为免疫球蛋白受体超家族成员,主要表达于破骨细胞、巨噬细胞和小胶质细胞,通过结合磷脂、APOE、Aβ寡聚体和凋亡神经元等配体激活Ca²⁺动员、MAPK(丝裂原活化蛋白激酶)和mTOR(哺乳动物雷帕霉素靶蛋白)等下游信号通路,在AD小鼠模型中,其通过表达于小胶质细胞中发挥神经保护作用,是神经退行性疾病中免疫反应的关键调控因子。研究显示,Trem2敲除(Trem2−/−)或携带TREM2 R47H突变会减少斑块相关小胶质细胞并增加营养不良性神经突起,加剧Aβ毒性及tau的播种和扩散;而TREM2激活抗体可增强小胶质细胞代谢与功能,减少淀粉样斑块形成。
TREM2可被ADAM10/17等α分泌酶在组氨酸157位点切割发生胞外域脱落,产生可溶性TREM2(sTREM2),其能促进小胶质细胞存活、斑块聚集,并增强Aβ清除和细胞因子生成。TREM2和sTREM2均显著影响BBB功能,TREM2功能缺失或突变会通过增加BBB通透性促使外周免疫细胞及炎症因子侵入CNS,加剧神经炎症和退行性进程。sTREM2作为CNS与外周免疫系统的潜在桥梁,其水平变化可反映CNS内炎症状态和神经退行性变严重程度。AD患者脑脊液和血浆中sTREM2升高与疾病早期和轻度认知障碍相关,且与记忆衰退、海马萎缩相关,并与脑脊液tau/p-tau水平呈正相关,表明其作为AD进展生物标志物的潜在应用价值。
5.CD22
CD22作为最初被定义为B细胞受体的分子,研究发现其在CNS内对小胶质细胞功能具有关键调控作用。小胶质细胞通过吞噬清除蛋白聚集体和细胞碎片维持CNS稳态,但其功能在衰老及多种伴随认知下降的神经退行性疾病中退化。研究表明,CD22是小胶质细胞吞噬功能的负调控因子,在衰老小胶质细胞中表达上调,其通过与α2,6-连接唾液酸的相互作用发挥抗吞噬效应,抑制小胶质细胞的吞噬作用,促进淀粉样斑块的形成和传播,从而加剧神经炎症和神经退行性变化。可溶性CD22(sCD22)可作为炎症和小胶质细胞功能障碍的标志物,在AD患者外周血中水平升高,与脑脊液Aβ42水平及Aβ42/Aβ40比值呈负相关,与脑内磷酸化tau水平和淀粉样蛋白负荷呈正相关,且血浆sCD22升高与认知衰退加速相关,提示其可能会加速各种神经退行性疾病的进展。
在外周,CD22通过与B细胞受体相互作用调节B细胞活化和免疫应答,AD患者中CD22的上调可能导致外周免疫系统失调,加剧CNS炎症。动物模型显示,抑制CD22活性可增强小胶质细胞的吞噬活性,改善髓鞘碎片、Aβ寡聚体和α-突触核蛋白纤维的清除,从而改善认知功能。因此,CD22及其相关通路有望成为干预神经退行性疾病的潜在治疗靶点,为延缓或阻止疾病进展提供新策略。
三、总结
本综述聚焦AD中神经-免疫双向调控机制,阐明BBB破坏通过促进外周免疫细胞(如CD8⁺ T细胞、CD4⁺ T细胞)浸润加剧神经炎症,而脑内局部产生的APOE4、B2M、TREM2等免疫介质与神经退行性变呈因果关联。脑膜作为免疫监视平台,其淋巴管功能异常与Aβ沉积相关,而增强其功能联合Aβ抗体治疗可促进Aβ清除;免疫衰老背景下,慢性Aβ暴露诱导T细胞异常活化,驱动中枢炎症级联反应。值得注意的是,CD8⁺ T细胞在tau与Aβ病理模型中作用存在异质性,提示需结合病理背景解析其功能。与其他神经退行性疾病类似,AD的蛋白错误折叠通过激活模式识别受体触发神经炎症,其中TREM2 R47H突变和HLA-DRB1变异等免疫风险基因可协同放大病理进程。基于上述机制,靶向神经免疫互作的治疗策略(如调节Treg活性、阻断PD-1等免疫检查点及增强脑膜淋巴清除)展现出早期干预潜力,而明确外周免疫细胞的阶段特异性作用、鉴定新型免疫标志物及优化APOE4/TREM2靶向干预,将成为突破治疗瓶颈的关键方向。
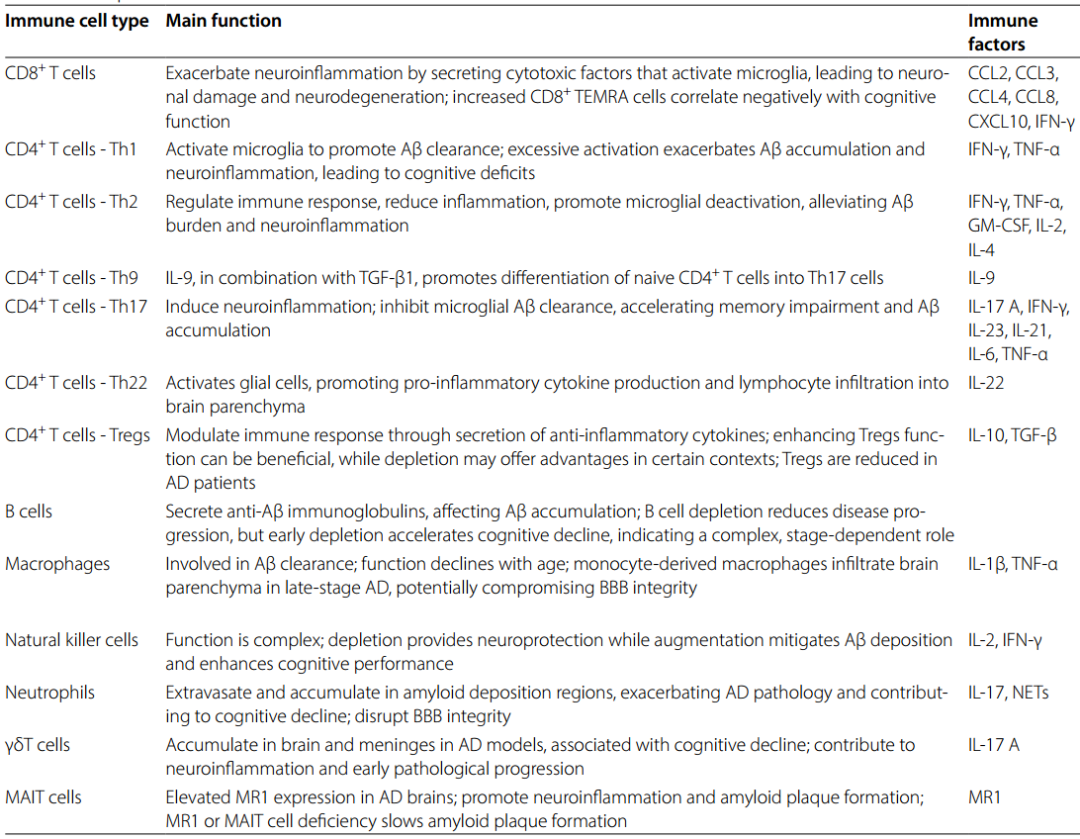
表1. AD患者外周免疫细胞的功能和机制

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK