2025-03-18 阅读量:18895
编者按
基因组编辑——即对生物遗传物质进行精准靶向修饰的技术,已成为分子生物学领域最具里程碑意义的突破之一。作为生命科学与医学研究的革命性工具,该技术不仅为解析复杂生命过程提供了全新视角,更推动了针对遗传性疾病根源的治疗策略发展。以CRISPR-Cas系统为代表的技术体系,凭借其高效性与高度可编程性,成为现代生物技术的核心驱动力。随着首款基于CRISPR的基因疗法(Casgevy)于2023年获美国FDA批准,基因组编辑技术正式迈入临床转化新阶段。
因此,小编在这里给大家推荐瑞士苏黎世大学Martin Jinek等人于2024年2月29日在国际生命科学领域顶刊Cell上发表的题为“Past, present, and future of CRISPR genome editing technologies”的综述文章,该文全面梳理CRISPR基因组编辑技术在基础研究与临床治疗中的发展现状与应用场景,重点解析制约其应用的技术瓶颈,并系统论述近年来为解决这些挑战而涌现的关键技术创新。
CRISPR基因组编辑技术的过去:发展历程与局限
1)技术演变
基因组编辑技术的起源可追溯至真核细胞DNA修复机制的研究进展。20世纪90年代初,研究者利用I-SceI等归巢核酸内切酶(可识别18 bp DNA序列)的开创性实验表明,在哺乳动物细胞中诱导靶向双链断裂(DSB)能促进靶向位点的同源重组,这奠定了基于DSB生成核酸酶的基因组编辑理论框架。由于真核细胞基因组靶向编辑需要具有长识别位点的多样化核酸酶以实现单一位点特异性切割,研究者随后开发了基于非特异性DNA内切酶与序列特异性DNA结合模块的串联序列融合的工程化核酸酶系统:如锌指核酸酶(ZFNs)及转录激活样效应因子核酸酶(TALENs),但繁琐的设计和生成过程限制了应用。直至2007年,原核细胞CRISPR-Cas系统被证实作为适应性基因组防御机制,可识别并靶向清除外源核酸(来自噬菌体)。该系统中,入侵DNA经转录和加工形成crRNA,其作为分子向导引导Cas蛋白识别并降解入侵核酸。2012年研究揭示:Cas9作为DNA切割核酸内切酶,其特异性由crRNA与反式激活crRNA(tracrRNA)共同构成的双RNA引导结构决定。通过将tracrRNA与crRNA整合为单链引导RNA(sgRNA),研究者进一步简化CRISPR-Cas9系统,实现了“一酶一RNA”的全可编程设计,只需设计具有匹配序列的sgRNA即可引导Cas9靶向特定基因组位点。2013年多项研究证实,在真核细胞中表达Cas9与特异性sgRNA可精准引入靶位点遗传修饰。
2)CRISPR核酸酶基因组编辑
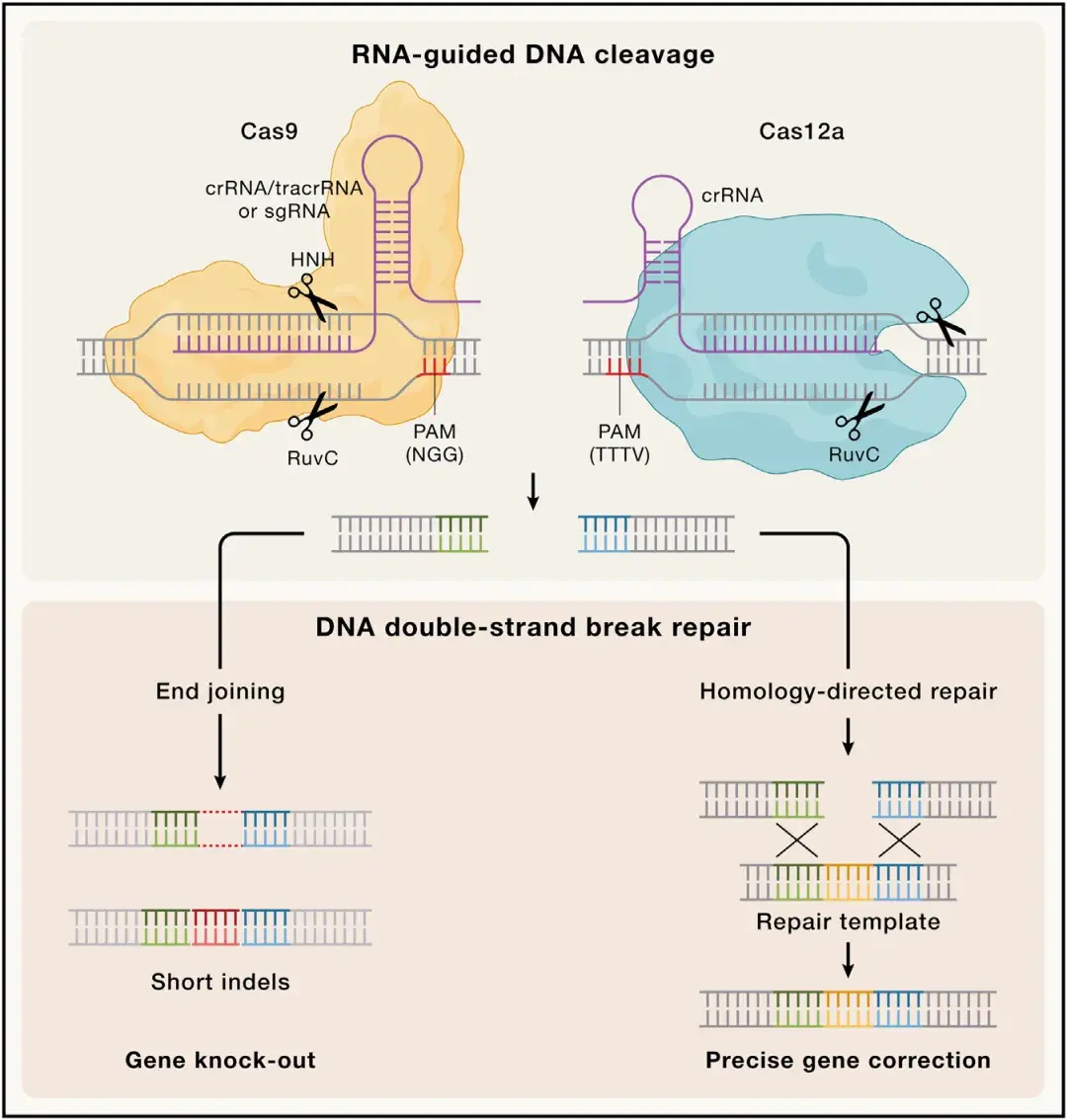
CRISPR-Cas核酸酶的可编程性使其能够产生位点特异性的双链DNA断裂,这一特性使其迅速应用于基因组编辑技术。源自化脓性链球菌(Streptococcus pyogenes)的典型Cas9蛋(SpCas9)作为首个被改造用于基因组编辑的Cas核酸酶,由于其固有的高活性和高特异性,至今仍是应用最广泛的基因编辑工具。为将Cas9核酸酶引导至目标基因组位点,可通过改变crRNA 5'端的20个核苷酸(20 nt)的引导序列,使其与DNA靶标进行标准碱基配对。靶向结合还依赖于DNA非靶链(non-target strand,NTS)上位于靶位点下游的短原间隔序列邻近基序(protospacer adjacent motif,PAM)。对PAM的识别会导致靶标DNA局部解链,随后引导RNA以5'→3'方向从PAM近端开始与DNA靶链(target strand,TS)进行碱基配对,引发Cas9构象变化从而激活核酸酶结构域。Cas9随后在PAM序列上游3个核苷酸处切割双链DNA(dsDNA),产生平末端或单核苷酸5'端突出的DSB。该切割由Cas9的HNH和RuvC结构域协同完成,其中HNH切割TS,RuvC切割NTS。
Cas12a源自V型CRISPR-Cas系统,与Cas9不同,Cas12a无需tracrRNA激活,而是通过识别crRNA重复序列中的保守假结结构实现核酸酶加工。Cas12a靶向具有5'端TTTV PAM的DNA,利用其单一RuvC催化位点在靶位点PAM远端依次切割两条链,产生5个核苷酸的5'端突出。研究表明Cas12a是一种高效核酸酶,具有与Cas9互补的特性和功能,可实现精准基因编辑。
传统基因组编辑技术依赖于在基因组中引入位点特异性双链DNA断裂,并通过内源性细胞DNA修复完成遗传修饰。Cas9或Cas12a产生的DSB通常通过易错的末端连接(end-joining)或精确的同源定向修复(HDR)机制修复。在哺乳动物细胞中,末端连接是主要修复方式,包括非同源末端连接(NHEJ)和微同源介导末端连接(MMEJ),通过直接连接断裂DNA末端实现。连接前对DNA末端的加工会导致核苷酸的添加或丢失,引起DSB位点处出现短的插入或缺失(统称为插入缺失indels)。研究表明,精准修复的DSB可能被反复切割,直到累积的indels阻止进一步切割,该特性被广泛用于通过邻近双DSB实现基因敲除或基因删除。相比之下,HDR是精确的DSB修复方式,需要同源DNA模板指导修复过程。通过外源提供人工同源修复模板(如质粒、病毒载体递送的dsDNA或合成的单链DNA寡核苷酸ssODN),HDR可在靶位点精准引入突变、插入或缺失。尽管HDR理论上可实现单碱基精度编辑,但由于修复因子通常只在细胞周期的S期和G2期表达,因此,HDR只在活跃分裂细胞中发生。HDR效率受修复模板类型、递送方法、细胞类型等因素影响,此外,需调控影响DNA修复方式选择的因素(如增强HDR并抑制末端连接)来优化编辑结果。
图1. CRISPR基因组编辑的分子原理
3)CRISPR基因组编辑的局限
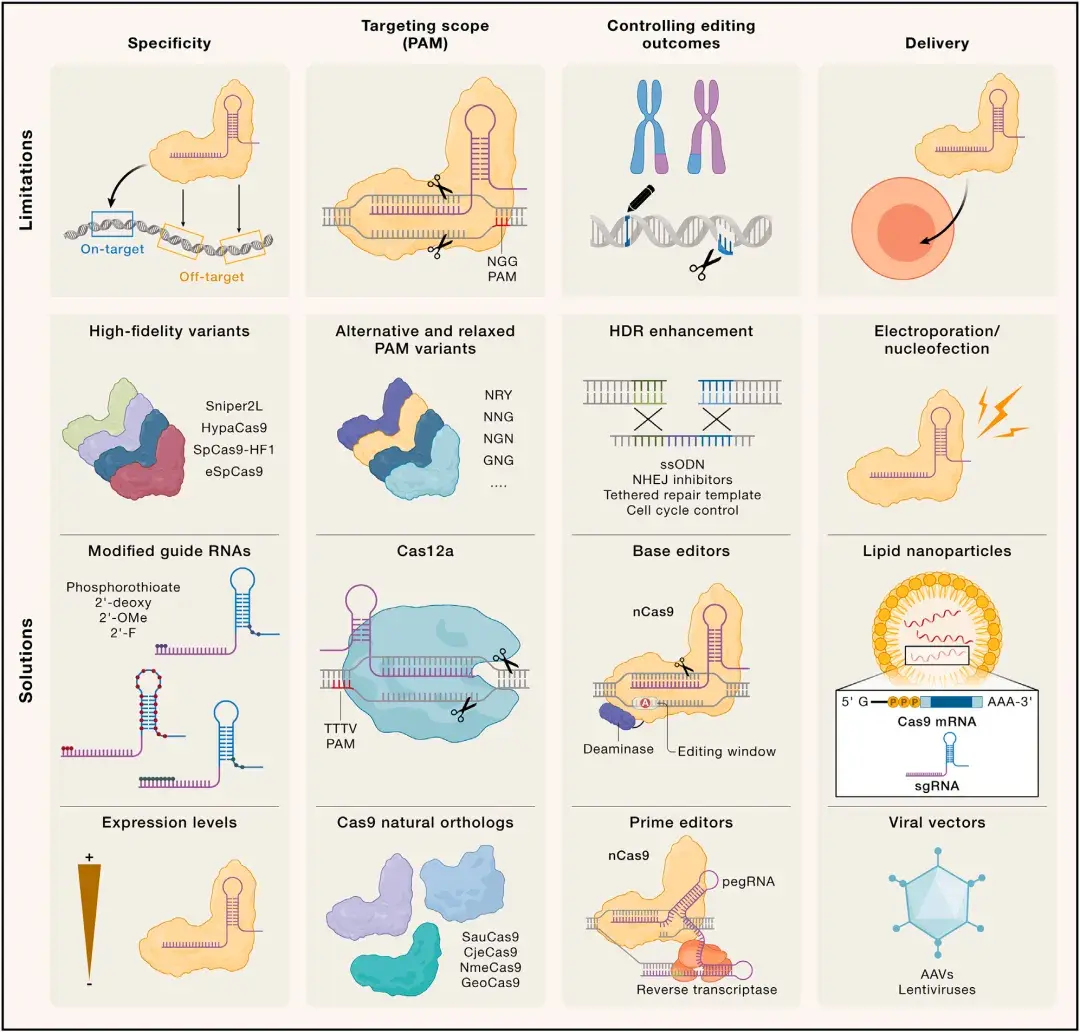
CRISPR-Cas系统被改造为简单高效的可编程基因编辑工具,极大推动了基础与应用研究的多领域发展,并为靶向基因疗法及各类生物技术应用奠定了基础。然而,这一高度进化的生物防御系统的功能特性与理想中精确基因组编辑工具的需求间存在差异。因此,第一代CRISPR基因编辑工具的应用潜力受到若干关键因素的限制,主要包括:特异性、靶向范围,及依赖内源性DSB修复机制实现基因组修饰。此外,CRISPR组件的递送也受限于递送载体和靶细胞。
脱靶效应
天然CRISPR-Cas系统对引导RNA与靶标DNA之间的错配具有一定容忍度,这可能是进化中为应对噬菌体高突变率而形成的特性。然而,它可能导致Cas9除了作用于预设靶位点外,还会靶向并编辑基因组中其它部分互补的脱靶位点。研究表明,Cas9对引导RNA-靶标DNA异源双链中的核苷酸错配(包括单个碱基错配、连续多碱基错配,甚至核苷酸插入或缺失)的耐受程度高度依赖引导序列本身。尽管Cas9具有错配容忍性,但其DNA结合与切割机制中的内在检查点使得大多数潜在脱靶位点仅被结合而不会引发双链DNA切割或编辑。此外,脱靶分析显示,与分离的基因组DNA相比,Cas9在体内的脱靶切割频率显著更低,这可能与基因组结构等因素对细胞中Cas9编辑活性的调控有关。尽管如此,基因组中多个位点同时发生脱靶切割仍可能导致基因组重排(如删除、倒位或染色体易位),并激活DNA损伤与应激响应通路。脱靶编辑仍是基因治疗应用的主要隐患,为此研究者致力于开发更灵敏的脱靶预测与检测方法,并通过分子工程手段(如高保真Cas9变体的设计)提升CRISPR编辑工具的特异性。
靶向范围受限(PAM依赖)
CRISPR核酸酶的DNA结合机制将其靶向范围限制于侧翼含PAM序列的基因组位点。尽管最常用的SpCas9核酸酶识别NGG型PAM(理论上平均每8个核苷酸存在一个潜在靶位点),但在高A/T含量的基因组区域仍难以找到合适靶点。Cas12a核酸酶识别富含T的PAM序列(如TTTV型),其靶向范围同样受限。
控制编辑结果
利用核酸酶在基因组内产生DSB可显著提高哺乳动物细胞的HDR效率。然而,HDR仅适用于分裂细胞,且由于末端连接方式的并行作用,常导致异源等位基因编辑结果。此外,靶位点的编辑精度还受其它不利因素限制,包括大片段删除、染色体重排甚至染色体丢失。因此,研究者致力于开发调控DNA修复结果的方法,尤其通过提升HDR效率以提高敲入(插入)突变的成功率。研究表明,修复模板的选择及其递送方式极大地影响HDR的效率。提高HDR效率的策略有:
1.使用非对称的ssODN模板。设计单链修复模板以优先引导HDR。
2.引入沉默突变。在靶位点引入不影响功能的突变,避免Cas9反复切割修复后的DNA。
3.将修复模板直接锚定至DSB位点。
4.细胞周期调控。同步化处理:通过小分子抑制剂暂缓S期进程或增加G2/M期细胞比例。
5.预组装Cas9-核糖核蛋白(RNP)递送:结合细胞周期同步化,提高HDR效率。
6.抑制NHEJ关键因子:如通过敲低Ku蛋白或抑制DNA-PKcs,抑制NHEJ以偏向HDR。
7.HDR相关修复因子偶联:将Cas9与HDR相关修复因子(如Rad51、Rad52或Mre11)直接偶联,显著提升敲入效率。
递送
靶向递送基因编辑工具仍是大多数体内与体外基因编辑应用的主要瓶颈。安全、特异且高效地将CRISPR组件递送至目标细胞是成功实现治疗性基因组编辑的前提。此外,CRISPR组分及其递送载体的免疫原性对体内治疗应用构成潜在风险。研究显示,人体内存在针对Cas9的预存抗体和反应性T细胞,而在非人灵长类疾病模型中,Cas9免疫反应可能导致疗效下降。针对预存免疫的应对策略包括:1.Cas9免疫原性改造:去除Cas9蛋白中的免疫原性表位。2.免疫调节:通过药物抑制免疫反应。3.短暂表达控制:限制Cas9表达时长以减少免疫暴露。
根据靶细胞类型,Cas9/Cas12a酶及其引导RNA可以多种形式递送。在大多数体外(即离体)培养细胞应用中,由于电穿孔(核转染)或脂质体介导的转染效率较高,因此仍然是最常用的递送方式。Cas9和引导RNA组分可作为RNA、质粒DNA或体外重组的RNP复合物进行递送。质粒介导的Cas9复合物长期表达会提高脱靶编辑频率和质粒随机整合概率。因此,在离体治疗应用中,RNP的瞬时递送已成为基因编辑的首选方法。对于许多模式生物生殖细胞的基因组编辑,通常通过显微注射或电穿孔递送Cas9 RNP或mRNA。在哺乳动物细胞中实现CRISPR-Cas9系统的体内递送通常依赖病毒载体。腺病毒、慢病毒和腺相关病毒(AAV)均可被改造,以将载体中的病毒基因替换为基因编辑模块。AAV因其低免疫原性、高转导效率和广泛的细胞嗜性,仍然是体内递送的首选载体。然而,AAV是相对较小的病毒,其载体容量有限(约4.7 kb)。因此,除非使用超紧凑型启动子,否则很难将编码SpCas9(4.2 kb)及其sgRNA(约100 nt)的基因包装到单个AAV载体。通过脂质纳米颗粒(LNP)等非病毒方法可将Cas9 mRNA与合成引导RNA复合物或体外重组的RNP递送到体内细胞。这类方法的优势在于相较于病毒载体具有更高的安全性和更低的免疫原性。LNP通过内吞作用进入细胞后,其内容物可能逃逸内体进入细胞核,也可能被溶酶体降解从而限制递送效率。
图2. CRISPR基因组编辑的局限
CRISPR基因组编辑技术的现状
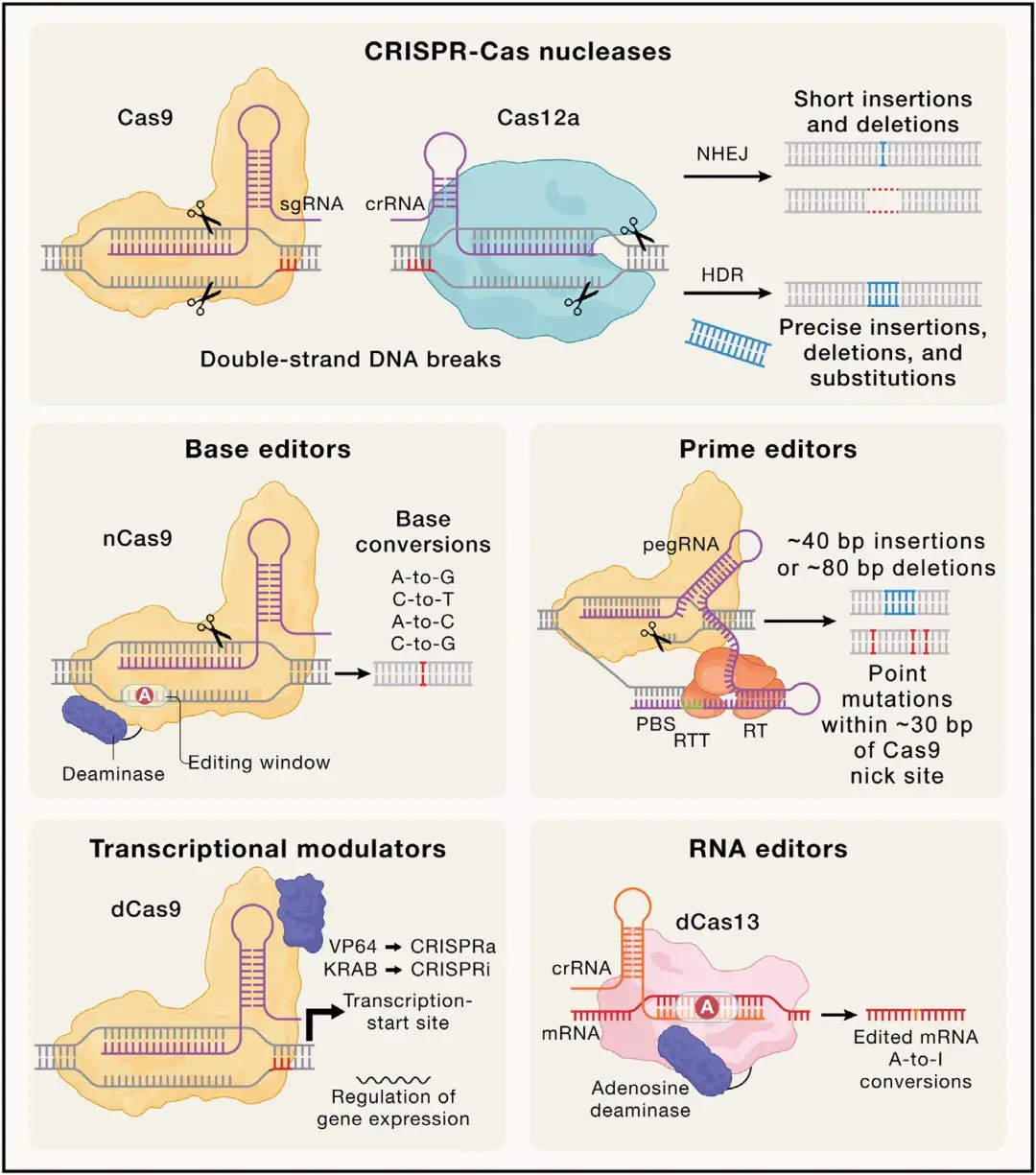
基于Cas9和Cas12a核酸酶的第一代依赖DSB的基因组编辑工具,通过持续创新不断提升了其功能——不仅增强了工具的通用性,还优化了其精准性并减少了非预期编辑后果。然而,关于其安全性的担忧依然存在,既包括脱靶编辑活性,也涉及靶向DSB的潜在基因毒性效应(如诱导p53反应)。对DSB基因毒性的担忧,以及解决HDR效率低下的需求,进一步推动了“第二代”CRISPR技术的发展——这些技术无需依赖DSB形成和HDR即可实现基因组编辑,主要包括碱基编辑(BEs)和先导编辑(PEs)。如图3所示,不同技术适用于特定类型的编辑或递送模式。
超越Cas9的核酸酶
近年来已有10多种在进化上多样化的RNA引导核酸酶被发现并应用于基因组编辑。2015年Cas12a的发现标志着Cas9之外的重大突破,它提供了具有独特PAM需求、引导RNA形式和DNA切割模式的新型核酸酶。据报道,Cas12a在体内表现出更高的特异性和更低的脱靶活性,部分归因于其较慢的DNA切割速率。此外,RNA靶向的RNA引导核酸酶(如Cas13、Cas12a2和Cas12g)为分子编辑工具箱增添了新维度,可实现mRNA的靶向降解或编辑以及核酸检测。
高保真Cas9变体
为解决脱靶活性问题,研究者通过两种互补策略开发了具有更高特异性的工程化SpCas9变体。第一种策略基于结构导向的理性设计,引入增强保真度的突变。其核心思路是消除Cas9蛋白与结合DNA靶点之间的特定接触,使Cas9-引导RNA复合物对底物DNA的错配更敏感,从而降低脱靶结合与切割的概率。针对这些变体的研究表明,突变显著减缓了DNA切割速率,进而促进脱靶位点的释放。第二种策略利用定向进化方法筛选能够减少脱靶编辑的突变。类似的努力也致力于设计其它Cas9和Cas12a酶的高保真突变体。
引导RNA修饰
对基因组编辑工具中引导RNA进行工程化改造以提高特异性,是替代高保真核酸酶突变体的另一有效策略。早期研究聚焦于使用截短型引导RNA(将Cas9的引导区从20个核苷酸缩短至17-18个核苷酸)。这类设计被证明可显著降低SpCas9在多种位点的脱靶活性,但同时也导致编辑效率下降或引发新脱靶位点的编辑。引导区5'端的修饰(如引入二级结构或未配对核苷酸)也被证实可抑制脱靶识别。多项研究发现引导区内2'-脱氧核苷酸替换可显著提高细胞内基因编辑的特异性。此外,在引导RNA中引入2'-O-甲基化、2'-氟代核苷酸及硫代磷酸酯键等化学修饰,也被证明是提高Cas9特异性和增强引导RNA稳定性的有效策略。
识别NRN PAM的Cas9变体
为克服Cas核酸酶对PAM依赖性靶位点的限制,并使CRISPR-Cas9系统能够作用于更广泛的基因组位点,近期研究成功开发出可识别NRN(N代表任意碱基;R代表嘌呤A/G)PAM的SpCas9变体。类似地,具有放宽PAM特异性的Cas12a变体也已被开发用于基因编辑应用。
碱基编辑
CRISPR衍生的碱基编辑(BEs)作为一种多功能技术已被开发,可在不产生DSB且无需提供同源重组修复模板的情况下实现靶向点突变,从而能够在同源重组缺陷(HDR-deficient)的细胞中进行编辑。碱基编辑由Cas9的RuvC结构域失活的切口酶版本与核苷酸脱氨酶模块化融合而成。最初开发了两类碱基编辑:胞嘧啶碱基编辑(CBEs)含有源自APOBEC1等胞苷脱氨酶的催化结构域及尿嘧啶糖基化酶抑制剂(UGI)结构域,可实现C-T的转换;而腺嘌呤碱基编辑(ABEs)则利用经定向进化改造的tRNA特异性脱氨酶TadA的腺苷脱氨酶结构域,使其能够作用于单链DNA,从而产生A-G的转换。当Cas9模块结合靶标后,碱基编辑会在非靶标DNA链的PAM远端区(即“编辑窗口”)内,将胞嘧啶或腺嘌呤分别脱氨基为尿嘧啶或肌苷(I)。这些修饰碱基在DNA复制过程中分别被识别为胸腺嘧啶和鸟嘌呤,从而诱导过渡型点突变。
自开发以来,初始CBE和ABE编辑已通过多次设计迭代来提升活性并减少脱氨酶诱导的脱靶编辑,同时基于Cas12a的碱基编辑也已被开发。碱基编辑的类型范围进一步扩展至A-C、A-Y和C-G等颠换突变。由于编辑结果具有高度可预测性,碱基编辑已被应用于全基因组敲除和突变筛选研究。其精准性使其适用于由单点突变引发疾病的治疗性校正。
先导编辑
先导编辑是一种基于Cas9的技术,旨在以不依赖HDR的方式产生靶向点突变以及插入或缺失。先导编辑由先导编辑引导RNA(pegRNA)和融合蛋白构成,该融合蛋白包含HNH结构域失活的Cas9切口酶及工程化逆转录酶(RT)结构域。pegRNA的3'端含有与基因组靶标NTS互补的序列延伸,其中包含所需突变。Cas9在NTS上产生切口后,该切口区域与pegRNA的延伸序列通过碱基配对结合。随后,逆转录酶以pegRNA为模板催化NTS的3'端延伸,从而引入突变。DNA链重新退火形成5'flap中间体,经切除与连接后完成基因组DNA编辑的固定。这种靶向链合成技术可实现最长约40 bp的插入、约80 bp的缺失,以及距离Cas9切口位点约30 bp范围内的点突变。从第一代先导编辑[将Cas9切口酶与莫洛尼鼠白血病病毒(MMLV)野生型RT融合]开始,后续改进版本通过整合热稳定性增强的工程化MMLV RT结构域,并引入第二个sgRNA在非编辑链产生切口以促进编辑保留,显著提升了编辑效率。此外,通过抑制DNA错配修复途径、优化核定位、提高表达水平及DNA切口活性,进一步实现了性能提升。在pegRNA的3'端添加稳定二级结构以抵抗降解的策略也被证明可提高编辑效率。迄今为止,先导编辑已成功应用于多种生物和细胞类型。
转录调控:CRISPRi和CRISPRa
CRISPR技术不仅可用于基因组编辑,还可实现对基因表达的瞬时调控。催化失活的Cas9突变体最初在细菌中被用于靶向基因启动子,通过空间位阻效应阻断RNA聚合酶,从而抑制RNA转录。为抑制真核细胞的基因表达,核酸酶失活的Cas9可与多种转录及表观遗传调控因子(如KRAB转录抑制结构域)融合,并靶向活跃转录基因的启动子区域。该方法被称为CRISPR干扰(CRISPRi),可作为小干扰RNA(siRNA)介导RNA干扰的替代方案实现高效基因表达敲降。类似地,核酸酶失活的Cas9蛋白可通过直接募集转录激活因子或调控染色质状态来激活特定基因表达。CRISPR激活(CRISPRa)通过将dCas9与VP64等转录激活结构域及其衍生物融合实现。此外,与组蛋白修饰酶(如去甲基化酶或乙酰化酶)的融合可位点特异性地改变特定位点的表观遗传标记,诱导活性染色质状态以驱动基因表达。此类方法已广泛应用于全基因组水平的功能缺失(CRISPRi)或功能获得(CRISPRa)筛选。然而,由于Cas9与DNA的结合通常比切割更具非特异性,CRISPR介导的转录调控存在显著的脱靶活性。
靶向RNA沉默与修饰
VI型CRISPR-Cas系统中RNA引导的单链RNA靶向Cas13酶的发现,推动了靶向RNA沉默、编辑及修饰工具的开发。由于RNA编辑具有瞬时性,在某些场景下转录组编辑可能成为基因组编辑的替代方案。例如,在疼痛、炎症或病毒感染等急性疾病中,瞬时RNA编辑可提供暂时的治疗效果,避免引入永.久性遗传改变。当前RNA编辑策略主要基于将核酸酶失活的dCas13蛋白与作用于RNA的腺苷脱氨酶(ADAR)融合,催化靶RNA中的腺苷脱氨基生成肌苷。引导RNA与靶RNA的杂交形成双链RNA底物供ADAR酶作用,产生的肌苷在翻译时可与胞嘧啶配对[被核糖体识别为鸟苷(G)],从而在mRNA中实现等效于A-G的密码子转换。研究人员通过蛋白工程改造,进一步开发出ADAR2变体,可实现靶RNA中C-U的转换。基于dCas13蛋白融合的靶向RNA修饰概念已拓展至表观转录组编辑领域,例如在靶向mRNA位点特异性引入N6-甲基腺苷(m6A)修饰。
图3. 当前CRISPR编辑技术
当前基础研究与医学领域的应用
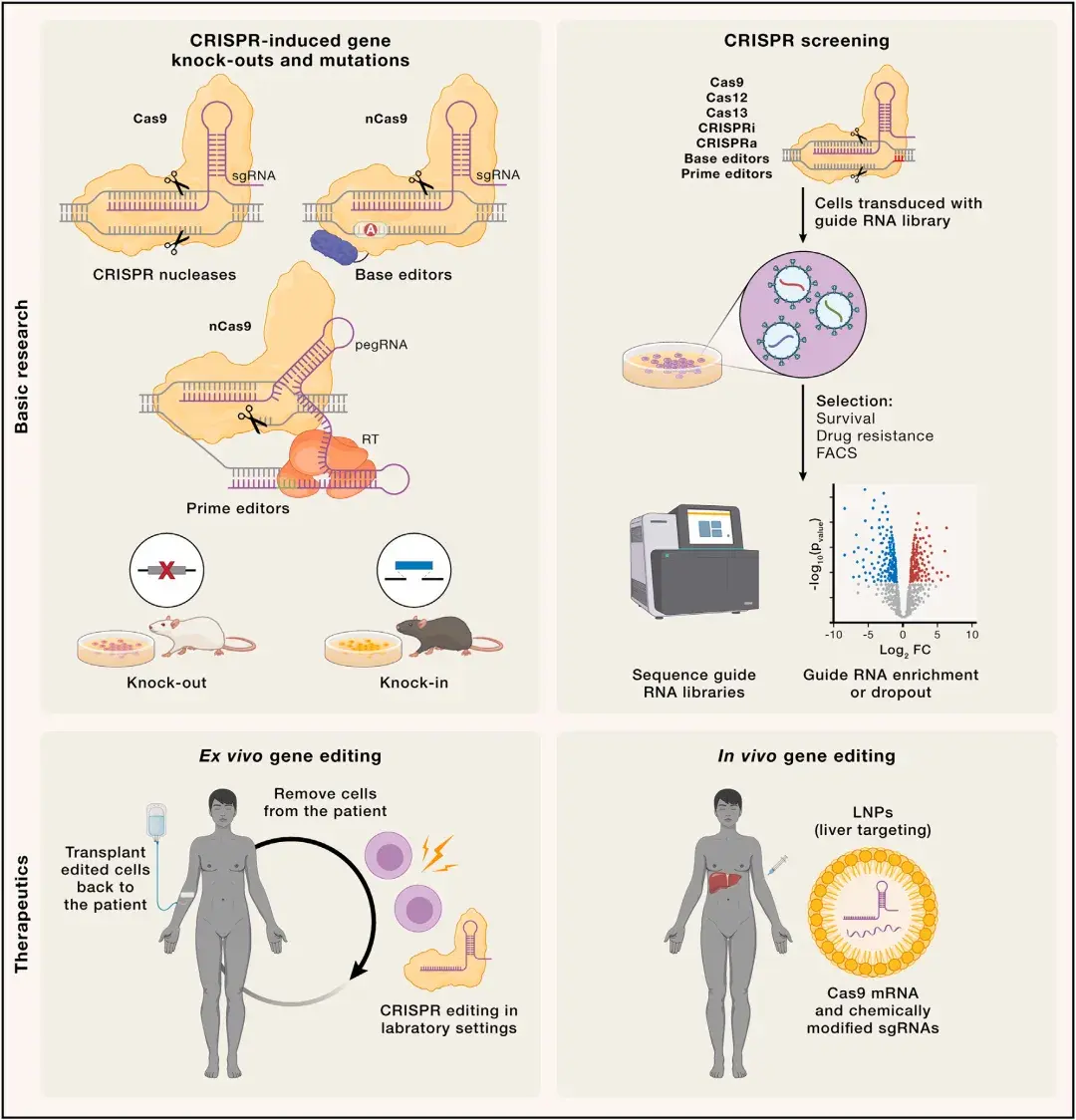
CRISPR基因组编辑技术的进步为提升人类健康水平开辟了广阔应用前景,涵盖基础研究突破与新型疗法开发。该技术促使科学家能够在多种实验模型中模拟致病突变、创建全基因组规模的高通量筛选方法。过去10年中,部分CRISPR治疗策略已从临床前研究推进至人体试验阶段,包括体内直接编辑与体外编辑回输两种路径。体内疗法通过递送编辑元件至靶组织实现基因校正,而体外疗法则通过编辑患者细胞(如造血干细胞或CAR-T细胞)后回输进行治疗。
反向遗传学:细胞与动物中疾病建模
鉴定人类疾病的致病等位基因是遗传研究的核心。CRISPR编辑技术通过构建等基因细胞模型(如在野生型细胞中引入特异突变或将患者细胞突变回复为野生型)或基因修饰动物模型,为验证致病变异提供了关键工具,因此,已广泛应用于患者细胞、模式生物的疾病表型建模。CRISPR编辑人诱导多能干细胞(iPSCs)并定向分化为疾病相关细胞类型,为研究难获取组织(如脑组织)的疾病机制提供有力工具。啮齿类(主要为小鼠)作为常用动物疾病模型,CRISPR技术可实现多基因或条件性敲除/敲入。CRISPR编辑非人灵长类等大动物模型因其生理和遗传特征接近人,成为研究衰老、慢性病与心血管疾病的理想平台。此外,除了验证疾病相关变异和理解疾病机制之外,啮齿类和大动物模型为疾病的治疗干预提供了重要的临床前评估平台。CRISPR技术无需维持大规模繁殖种群即构建大动物模型,具有显著的经济效益和动物福利优势。该技术还能通过使用不同引导RNA同时靶向多个基因组位点实现多重编辑。这一策略已成功用于构建携带多基因突变的动物模型。
正向遗传学:CRISPR筛选
全基因组敲除筛选等正向遗传学方法是研究生物学功能的重要工具。CRISPR筛选通过系统扰动数千基因/非编码元件,鉴定特定通路相关基因及其互作。高通量筛选中,通过转导慢病毒载体递送引导RNA文库与Cas酶至细胞(以低感染复数感染细胞,确保每个细胞仅整合一种sgRNA),经表型筛选后通过二代测序分析引导RNA富集情况,从而识别与特定表型相关的靶基因。该策略已拓展至CRISPRi、CRISPRa、碱基编辑、先导编辑及Cas13等多类型CRISPR技术。早期筛选聚焦细胞增殖/耐药等易选表型相关的基因,多重CRISPR筛选可发现抑制癌细胞生长的成对基因互作。碱基编辑能有效引入点突变,已成为功能获得/缺失型遗传变异筛选的强效工具。
体外治疗应用
提取患者细胞经CRISPR编辑后扩增并回输体内,已成为治疗遗传性血液病的重要策略。如首例遗传性血红蛋白病相关疗法已获欧洲、英国和美国监管机构批准。镰状细胞病(SCD)和输血依赖性β-地中海贫血(TDT)由血红蛋白β亚基(HBB)的多种突变引发。胎儿γ-血红蛋白(HBG)表达通常在出生后被转录抑制因子BCL11A沉默,研究者利用Cas9 RNP电穿孔靶向破坏CD34+造血干细胞中BCL11A基因的红系增强子区域,从而重新激活HBG表达,已成功用于SCD和TDT治疗。
嵌合抗原受体(CAR)T细胞疗法通过体外编辑T细胞,使其靶向癌细胞特定标志物,如B细胞恶性肿瘤中的CD19或多发性骨髓瘤中的B细胞成熟抗原(BCMA),从而在回输患者体内后清除癌细胞。现行获批的自体CAR-T疗法中,CAR编码基因通过慢病毒载体随机整合至基因组,且细胞制备过程复杂易失败。为突破这些局限,利用CRISPR编辑供体来源T细胞制备同种异体CAR-T细胞,可显著提升功能特性、简化生产工艺并优化质量控制。通过HDR将CAR编码基因精准插入T细胞受体α恒定区(TRAC)位点,可避免移植物抗宿主病;而敲除PDCD1、Regnase-1和TGFBR2基因则旨在减少T细胞耗竭并延长其体内存续时间。目前多项针对血液癌症(如白血病、淋巴瘤、骨髓瘤)的CRISPR编辑CAR-T疗法临床试验正在进行,其中突破性案例包括使用多重碱基编辑的CAR-T细胞治疗儿童复发型T细胞白血病——这一既往难以治愈的疾病。
体内治疗应用
与体外策略不同,体内治疗性基因编辑需要通过局部或全身递送方式,将CRISPR基因组编辑工具直接递送到体内靶组织。由于肝脏具有不连续的毛细血管结构,可通过低密度脂蛋白(LDL)受体途径摄取全身给药的脂质纳米颗粒,因此它是体内基因编辑的常用靶标。转甲状腺素蛋白(TTR)淀粉样变性是一种由心脏或神经系统中有害的错误折叠TTR积累引起的疾病。由于TTR主要在肝脏中产生,针对TTR淀粉样变性的一种有效治疗策略是通过LNP介导的递送,将Cas9 mRNA和合成sgRNA递送到肝脏中敲除该基因。在一项临床试验中,这种方法显著降低了血清TTR水平,成为首个系统性递送的体内CRISPR基因治疗成功案例。
图4. CRISPR基因组编辑技术在人类健康相关领域的应用
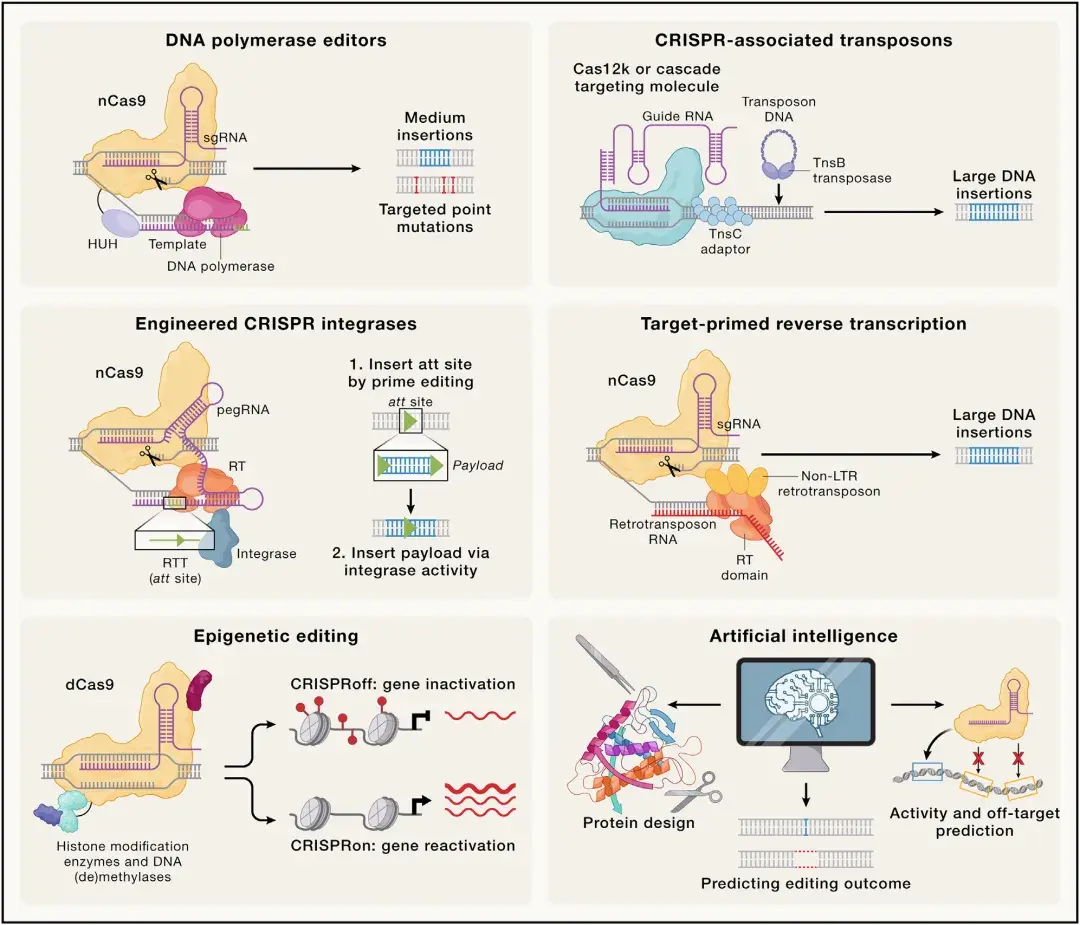
CRISPR基因组编辑技术的未来:新兴技术
随着当前CRISPR技术在过去10年间的局限日益显现,为突破这些限制并提升CRISPR基因组编辑的效率与通用性,科研人员成功开发了新兴的第三代工具和技术。
CAST系统
转座子能够在不依赖DSB生成与修复的条件下自主催化大片段DNA的插入。多项研究尝试将核酸酶失活的Cas9与不同转座酶(如睡美人转座子、mariner转座子及piggyBac转座子)融合,以实现RNA介导的细胞内转座。尽管这些策略能提高靶位点附近的转座事件频率,但整体效率低下及高脱靶转座频率限制了其作为基因插入技术的广泛应用。与工程化的Cas9-转座酶融合系统不同,CRISPR相关转座(CAST)系统是天然存在的Tn7样转座元件,其通过整合I型或V型CRISPR-Cas系统作为靶向模块,实现RNA引导的DNA转座。该系统能在细菌中介导高效、定点插入。尽管V型CAST在人细胞中的效率仍较低,但I型CAST活性较高,其作为大片段基因定点插入技术具有潜力。
逆转录元件编辑
与CAST系统不同,以非长末端重复(non-LTR)逆转录转座子为代表的逆转录元件,为长片段DNA序列的可编程插入提供了具有更高效率和特异性的新兴技术路径。这类元件通过靶向引发逆转录(TPRT)机制催化DNA插入:首先在靶DNA产生切口,利用切口暴露的3'末端作为引物启动逆转录转座子RNA的逆转录过程,生成互补DNA(cDNA)并通过宿主修复机制整合到基因组中。科学家进一步将逆转录转座子的DNA识别模块替换为Cas9切口酶;利用逆转录转座子的逆转录酶活性,以切口处的3'端为引物,将外源RNA模板(携带需插入的基因序列)反转录为DNA,并整合到基因组中。
表观基因组编辑
永.久性基因编辑(引入永.久遗传修饰)可能引发意外突变并涉及伦理争议,而CRISPR表观基因组编辑技术——通过引入靶向表观修饰实现不改变DNA序列的持续基因表达调控——为此提供了替代方案。将表观修饰酶与核酸酶失活的Cas9融合,可在特定基因组位点重构染色质状态,实现基因表达的长期激活或抑制。表观基因组编辑技术中CRISPRoff系统通过联合DNA甲基转移酶如Dnmt3A/Dnmt3L与KRAB转录抑制结构域,可在增殖细胞中介导高效持久的基因沉默;其效应可通过CRISPRon系统(整合DNA去甲基酶TET1与转录激活结构域,重新激活基因表达)实现逆转。组蛋白乙酰化修饰方面,组蛋白乙酰转移酶HATs和去乙酰化酶HDACs可分别用于染色质修饰以激活或抑制基因表达。
图5. 基因组编辑中的新兴技术
新递送方法
基因编辑复合物向靶细胞或器官的高效精准递送仍是基因组编辑技术面临的核心瓶颈。当前,多种策略正在开发中以提升递送效率并降低CRISPR编辑工具的潜在免疫原性。新型LNP递送系统通过优化脂质配方实现了组织特异性靶向,为CRISPR组件的组装与递送提供了通用平台,其细胞摄取效率显著提升且脱靶编辑风险降低。细胞穿透肽作为载体递送CRISPR核酸酶在神经元细胞编辑中展现出潜力。经工程化改造的细菌收缩注射系统可实现蛋白(如基因组编辑核酸酶)的瞬时、细胞特异性高效递送。病毒样颗粒正在成为体内应用中替代传统病毒载体的有力选择。
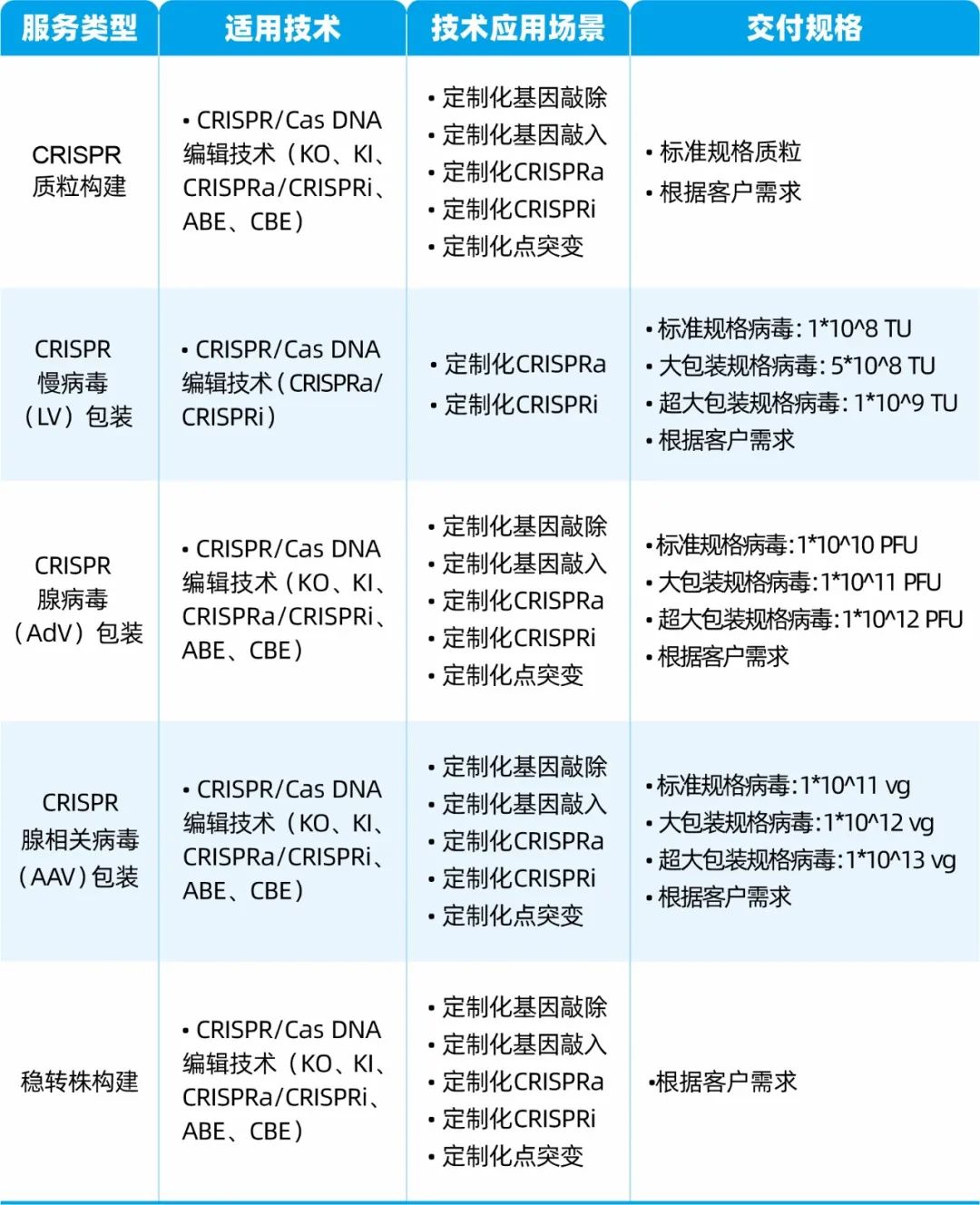
目前,枢密科技已开发CRISPR/Cas DNA编辑系列病毒载体,提供现货产品、定制病毒包装及细胞系构建服务,相关内容如下:

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK