Survivin(Mix)Mouse Monoclonal Antibody
产品基本信息
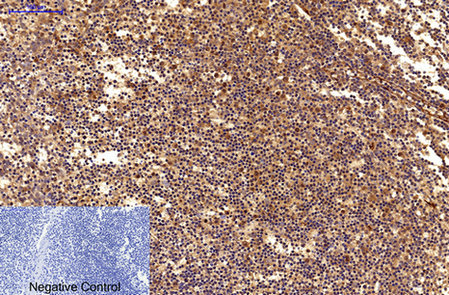
Immunohistochemical analysis of paraffin-embedded Human-Tonsil tissue. 1,Survivin Monoclonal Antibody(Mix) was diluted at 1:200(4°C,overnight). 2, Sodium citrate pH 6.0 was used for antibody retrieval(>98°C,20min). 3,Secondary antibody was diluted at 1:200(room tempeRature, 30min). Negative control was used by secondary antibody only.
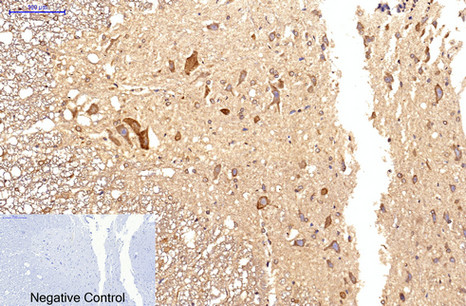
Immunohistochemical analysis of paraffin-embedded Rat-spinal-cord tissue. 1,Survivin Monoclonal Antibody(Mix) was diluted at 1:200(4°C,overnight). 2, Sodium citrate pH 6.0 was used for antibody retrieval(>98°C,20min). 3,Secondary antibody was diluted at 1:200(room tempeRature, 30min). Negative control was used by secondary antibody only.
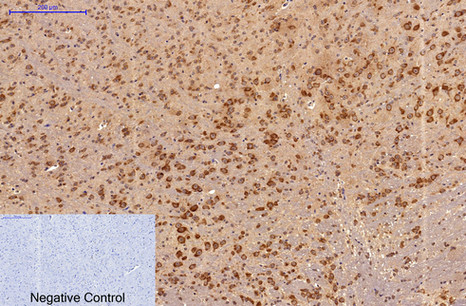
Immunohistochemical analysis of paraffin-embedded Mouse-brain tissue. 1,Survivin Monoclonal Antibody(Mix) was diluted at 1:200(4°C,overnight). 2, Sodium citrate pH 6.0 was used for antibody retrieval(>98°C,20min). 3,Secondary antibody was diluted at 1:200(room tempeRature, 30min). Negative control was used by secondary antibody only.
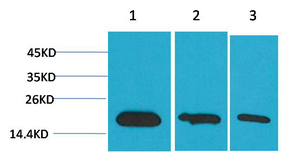
Western blot analysis of 1) Hela, 2) 293, 3) PC12 using Survivin Monoclonal Antibody.

Immunohistochemical analysis of paraffin-embedded human colon caricnoma using Survivin Monoclonal Antibody.
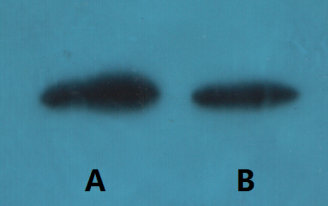
Western blot detection of Survivin in human breast cancer cell line MCF-7(A) and Cal51 (B) using Survivin mouse mAb (1:1000 diluted).Predicted band size: 16kDa.Observed band size:16kDa. Picture was kindly provided by our customer from Tianjin Medical University Cancer Institute and Hospital
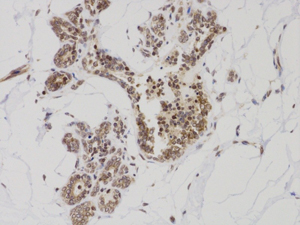
Immunohistochemical analysis of paraffin-embedded Human breast cancer. 1, Using Survivin Mouse mAb diluted at 1:200 (4°,overnight). 2, High-pressure and temperature Citric acid, pH6.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 50min). Picture was kindly provided by our customer from Tianjin Medical University Cancer Institute and Hospital
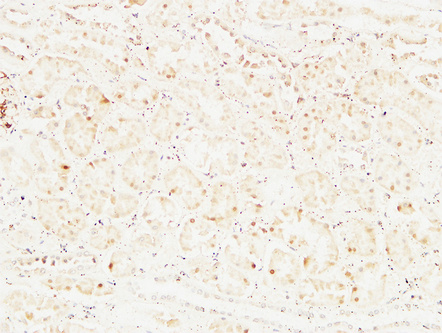
Immunohistochemical analysis of paraffin-embedded Human kidney. 1, Antibody was diluted at 1:200(4°,overnight). 2, High-pressure and temperature EDTA, pH8.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 30min).
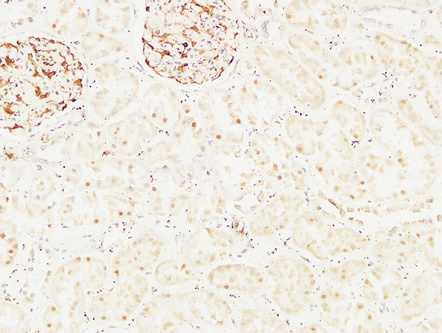
Immunohistochemical analysis of paraffin-embedded Human kidney. 1, Antibody was diluted at 1:200(4°,overnight). 2, High-pressure and temperature EDTA, pH8.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 30min).
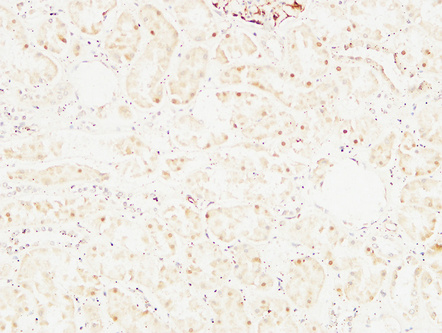
Immunohistochemical analysis of paraffin-embedded Human kidney. 1, Antibody was diluted at 1:200(4°,overnight). 2, High-pressure and temperature EDTA, pH8.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 30min).
相关文献
产品问答
相关产品

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK
