产品名称
SDHA Rabbit Polyclonal Antibody
别名
SDHA; SDH2; SDHF; Succinate dehydrogenase [ubiquinone] flavoprotein subunit; mitochondrial; Flavoprotein subunit of complex II; Fp
蛋白名称
Succinate dehydrogenase [ubiquinone] flavoprotein subunit mitochondrial
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=6389
Human Swissprot No.
P31040
Human Swissprot Link
http://www.uniprot.org/uniprotkb/P31040/entry
Mouse Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=66945
Mouse Swissprot No.
Q8K2B3
Mouse Swissprot Link
http://www.uniprot.org/uniprot/Q8K2B3
Rat Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=157074
Rat Swissprot Link
http://www.uniprot.org/uniprot/Q920L2
免疫原
The antiserum was produced against synthesized peptide derived from human SDHA. AA range:551-600
特异性
SDHA Polyclonal Antibody detects endogenous levels of SDHA protein.
宿主
Polyclonal, Rabbit,IgG
背景介绍
This gene encodes a major catalytic subunit of succinate-ubiquinone oxidoreductase, a complex of the mitochondrial respiratory chain. The complex is composed of four nuclear-encoded subunits and is localized in the mitochondrial inner membrane. Mutations in this gene have been associated with a form of mitochondrial respiratory chain deficiency known as Leigh Syndrome. A pseudogene has been identified on chromosome 3q29. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2014],
组织表达
Adipocyte,Brain,Colon,Heart,Liver,Placenta,
细胞定位
Mitochondrion inner membrane ; Peripheral membrane protein ; Matrix side .
信号通路
Citrate cycle (TCA cycle);Oxidative phosphorylation;Alzheimer's disease;Parkinson's disease;Huntington's disease;
功能
catalytic activity:Succinate + ubiquinone = fumarate + ubiquinol.,cofactor:FAD.,disease:Defects in SDHA are a cause of complex II mitochondrial respiratory chain deficiency [MIM:252011]; also known as succinate CoQ reductase deficiency. Defects of oxidative phosphorylation give rise to heterogeneous clinical symptoms ranging from isolated organ dysfunction to multisystem disorder. A deficiency of complex II represents a rare cause of mitochondrial encephalomyopathy, leukodystrophy, late-onset optic atrophy and ataxia, myopathy with exercise intolerance, and isolated cardiomyopathy.,disease:Defects in SDHA are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,function:Flavoprotein (FP) subunit of succinate dehydrogenase (SDH) that is involved in complex II of the mitochondrial electron transport chain and is responsible for transferring electrons from succinate to ubiquinone (coenzyme Q).,miscellaneous:The complex, present in mitochondria, can be degraded to form EC 1.3.99.1, which no longer reacts with ubiquinone.,pathway:Carbohydrate metabolism; tricarboxylic acid cycle.,sequence caution:Differs extensively from that shown.,similarity:Belongs to the FAD-dependent oxidoreductase 2 family. FRD/SDH subfamily.,subunit:Component of complex II composed of four subunits: the flavoprotein (FP) sdha, iron-sulfur protein (IP) sdhb, and a cytochrome b560 composed of sdhc and sdhd.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
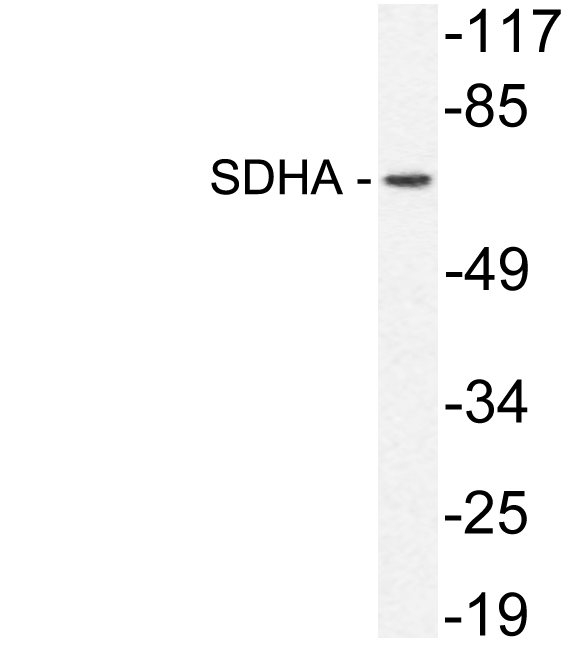
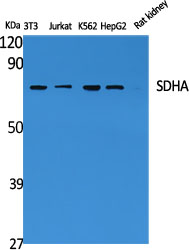
.jpg)

