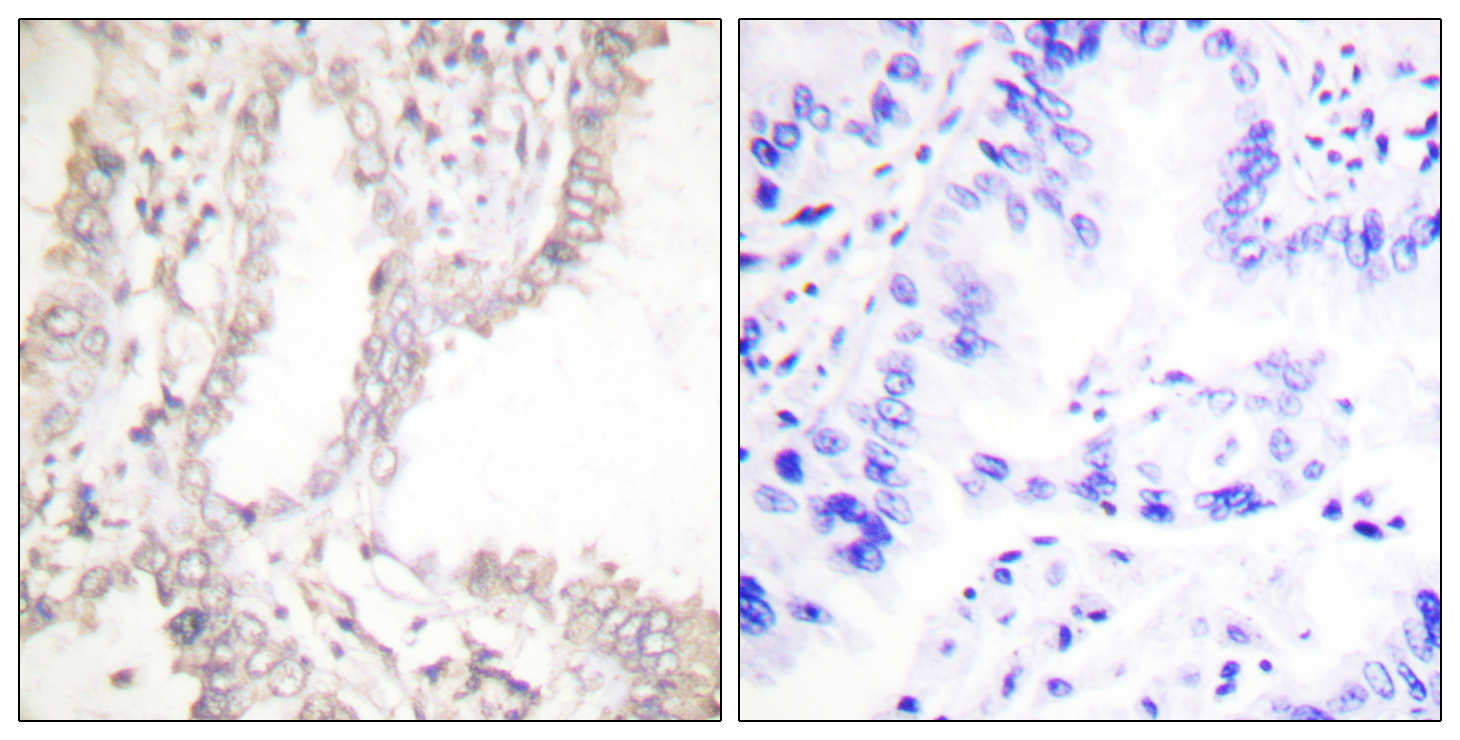
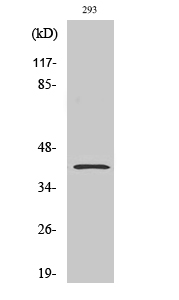
产品名称
PAR4 Rabbit Polyclonal Antibody
别名
PAWR; PAR4; PRKC apoptosis WT1 regulator protein; Prostate apoptosis response 4 protein; Par-4
蛋白名称
PRKC apoptosis WT1 regulator protein
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=5074
Human Swissprot No.
Q96IZ0
Human Swissprot Link
http://www.uniprot.org/uniprotkb/Q96IZ0/entry
Mouse Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=114774
Mouse Swissprot No.
Q925B0
Mouse Swissprot Link
http://www.uniprot.org/uniprot/Q925B0
Rat Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=64513
Rat Swissprot Link
http://www.uniprot.org/uniprot/Q62627
免疫原
The antiserum was produced against synthesized peptide derived from human Prostate Apoptosis Response protein-4. AA range:291-340
特异性
PAR4 Polyclonal Antibody detects endogenous levels of PAR4 protein.
宿主
Polyclonal, Rabbit,IgG
背景介绍
The tumor suppressor WT1 represses and activates transcription. The protein encoded by this gene is a WT1-interacting protein that itself functions as a transcriptional repressor. It contains a putative leucine zipper domain which interacts with the zinc finger DNA binding domain of WT1. This protein is specifically upregulated during apoptosis of prostate cells. [provided by RefSeq, Jul 2008],
组织表达
Widely expressed. Expression is elevated in various neurodegenerative diseases such as amyotrophic lateral sclerosis, Alzheimer, Parkinson and Huntington diseases and stroke. Down-regulated in several cancers.
细胞定位
Cytoplasm. Nucleus. Mainly cytoplasmic in absence of apoptosis signal and in normal cells. Nuclear in most cancer cell lines. Nuclear entry seems to be essential but not sufficient for apoptosis (By similarity). Nuclear localization includes nucleoplasm and PML nuclear bodies. .
功能
domain:The leucine-zipper domain is not essential for apoptosis, but is required for sensitization of cells to exogenous apoptotic insults and for interaction with its partners.,domain:The SAC domain is a death-inducing domain selective for apoptosis induction in cancer cells. This domain is essential for nuclear entry, Fas activation, inhibition of NF-kappa-B activity and induction of apoptosis in cancer cells.,function:Pro-apoptopic protein capable of selectively inducing apoptosis in cancer cells, sensitizing the cells to diverse apoptotic stimuli and causing regression of tumors in animal models. Induces apoptosis in certain cancer cells by activation of the Fas prodeath pathway and coparallel inhibition of NF-kappa-B transcriptional activity. Inhibits the transcriptional activation and augments the transcriptional repression mediated by WT1. Down-regulates the anti-apoptotic protein BCL2 via its interaction with WT1. Seems also to be a transcriptional repressor by itself. May be directly involved in regulating the amyloid precursor protein (APP) cleavage activity of BACE1.,induction:By apoptosis.,PTM:Preferentially phosphorylated at the Thr-163 by PKC in cancer cells.,subcellular location:Mainly cytoplasmic in absence of apoptosis signal and in normal cells. Nuclear in most cancer cell lines. Nuclear entry seems to be essential but not sufficient for apoptosis (By similarity). Nuclear localization includes nucleoplasm and PML nuclear bodies.,subunit:Interacts with WT1, via the C-terminal region. Homooligomer. Interacts also with a wide variety of proteins, such as atypical PKCs, p62, DAPK3 kinase and THAP1. Interacts with actin, AATF, BACE1, SPSB1, SPSB2 AND SPSB4. Component of a ternary complex composed of SQSTM1 and PRKCZ.,tissue specificity:Widely expressed. Expression is elevated in various neurodegenerative diseases such as amyotrophic lateral sclerosis, Alzheimer, Parkinson and Huntington diseases and stroke. Down-regulated in several cancers.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.