产品名称
Cleaved-MMP-12 (G106) Rabbit Polyclonal Antibody
别名
MMP12; HME; Macrophage metalloelastase; MME; Macrophage elastase; ME; hME; Matrix metalloproteinase-12; MMP-12
蛋白名称
Macrophage metalloelastase
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=4321
Human Swissprot No.
P39900
Human Swissprot Link
http://www.uniprot.org/uniprotkb/P39900/entry
Mouse Swissprot No.
P34960
Mouse Swissprot Link
http://www.uniprot.org/uniprot/P34960
免疫原
The antiserum was produced against synthesized peptide derived from human MMP12. AA range:87-136
特异性
Cleaved-MMP-12 (G106) Polyclonal Antibody detects endogenous levels of fragment of activated MMP-12 protein resulting from cleavage adjacent to G106.
宿主
Polyclonal, Rabbit,IgG
背景介绍
This gene encodes a member of the peptidase M10 family of matrix metalloproteinases (MMPs). Proteins in this family are involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling, as well as in disease processes, such as arthritis and metastasis. The encoded preproprotein is proteolytically processed to generate the mature protease. This protease degrades soluble and insoluble elastin. This gene may play a role in aneurysm formation and mutations in this gene are associated with lung function and chronic obstructive pulmonary disease (COPD). This gene is part of a cluster of MMP genes on chromosome 11. [provided by RefSeq, Jan 2016],
组织表达
Found in alveolar macrophages but not in peripheral blood monocytes.
细胞定位
Secreted, extracellular space, extracellular matrix .
功能
catalytic activity:Hydrolysis of soluble and insoluble elastin. Specific cleavages are also produced at 14-Ala-|-Leu-15 and 16-Tyr-|-Leu-17 in the B chain of insulin.,cofactor:Binds 2 zinc ions per subunit.,cofactor:Binds 4 calcium ions per subunit.,domain:The conserved cysteine present in the cysteine-switch motif binds the catalytic zinc ion, thus inhibiting the enzyme. The dissociation of the cysteine from the zinc ion upon the activation-peptide release activates the enzyme.,function:May be involved in tissue injury and remodeling. Has significant elastolytic activity. Can accept large and small amino acids at the P1' site, but has a preference for leucine. Aromatic or hydrophobic residues are preferred at the P1 site, with small hydrophobic residues (preferably alanine) occupying P3.,induction:By exposure to lipopolysaccharide. Inhibited by dexamethasone.,similarity:Belongs to the peptidase M10A family.,similarity:Contains 4 hemopexin-like domains.,tissue specificity:Found in alveolar macrophages but not in peripheral blood monocytes.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
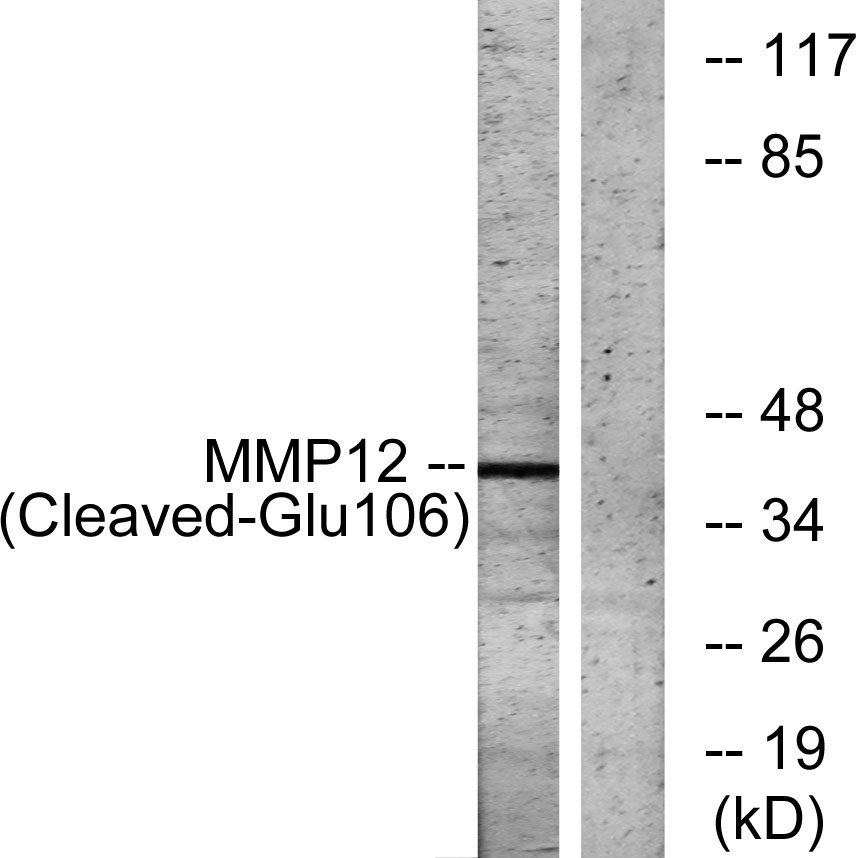
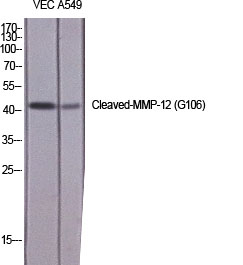
.jpg)

