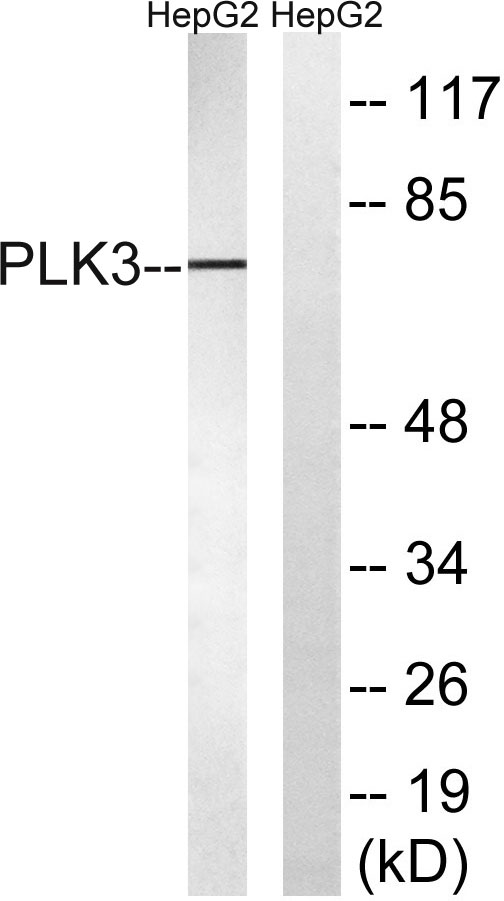
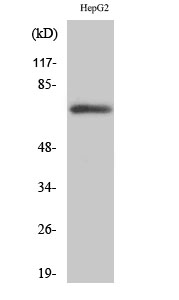
产品名称
Fnk Rabbit Polyclonal Antibody
别名
PLK3; CNK; FNK; PRK; Serine/threonine-protein kinase PLK3; Cytokine-inducible serine/threonine-protein kinase; FGF-inducible kinase; Polo-like kinase 3; PLK-3; Proliferation-related kinase
蛋白名称
Serine/threonine-protein kinase PLK3
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=1263
Human Swissprot No.
Q9H4B4
Human Swissprot Link
http://www.uniprot.org/uniprotkb/Q9H4B4/entry
Mouse Swissprot No.
Q60806
Mouse Swissprot Link
http://www.uniprot.org/uniprot/Q60806
Rat Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=58936
Rat Swissprot Link
http://www.uniprot.org/uniprot/Q9R011
免疫原
The antiserum was produced against synthesized peptide derived from human PLK3. AA range:231-280
特异性
Fnk Polyclonal Antibody detects endogenous levels of Fnk protein.
宿主
Polyclonal, Rabbit,IgG
背景介绍
The protein encoded by this gene is a member of the highly conserved polo-like kinase family of serine/threonine kinases. Members of this family are characterized by an amino-terminal kinase domain and a carboxy-terminal bipartite polo box domain that functions as a substrate-binding motif and a cellular localization signal. Polo-like kinases are important regulators of cell cycle progression. This gene has also been implicated in stress responses and double-strand break repair. In human cell lines, this protein is reported to associate with centrosomes in a microtubule-dependent manner, and during mitosis, the protein becomes localized to the mitotic apparatus. Expression of a kinase-defective mutant results in abnormal cell morphology caused by changes in microtubule dynamics and mitotic arrest followed by apoptosis. [provided by RefSeq, Sep 2015],
组织表达
Transcripts are highly detected in placenta, lung, followed by skeletal muscle, heart, pancreas, ovaries and kidney and weakly detected in liver and brain. May have a short half-live. In cells of hematopoietic origin, strongly and exclusively detected in terminally differentiated macrophages. Transcript expression appears to be down-regulated in primary lung tumor.
细胞定位
Cytoplasm. Nucleus. Nucleus, nucleolus. Golgi apparatus. Cytoplasm, cytoskeleton, microtubule organizing center, centrosome. Translocates to the nucleus upon cisplatin treatment. Localizes to the Golgi apparatus during interphase. According to a report, PLK3 localizes only in the nucleolus and not in the centrosome, or in any other location in the cytoplasm (PubMed:17264206). The discrepancies in results may be explained by the PLK3 antibody specificity, by cell line-specific expression or post-translational modifications. .
功能
catalytic activity:ATP + a protein = ADP + a phosphoprotein.,function:Serine/threonine protein kinase involved in regulating M phase functions during the cell cycle. May also be part of the signaling network controlling cellular adhesion. In vitro, is able to phosphorylate CDC25C and casein.,induction:Cytokine and cellular adhesion trigger FNK induction.,PTM:Phosphorylated as cells enter mitosis and dephosphorylated as cells exit mitosis.,similarity:Belongs to the protein kinase superfamily.,similarity:Belongs to the protein kinase superfamily. Ser/Thr protein kinase family. CDC5/Polo subfamily.,similarity:Contains 1 protein kinase domain.,similarity:Contains 2 POLO box domains.,subunit:Binds to the calcium/integrin-binding protein (CIB). This interaction probably occurs via the POLO-box domain.,tissue specificity:Transcripts are highly detected in placenta, lung, followed by skeletal muscle, heart, pancreas, ovaries and kidney and weakly detected in liver and brain. May have a short half-live. In cells of hematopoietic origin, strongly and exclusively detected in terminally differentiated macrophages. Transcript expression appears to be down-regulated in primary lung tumor.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.