产品名称
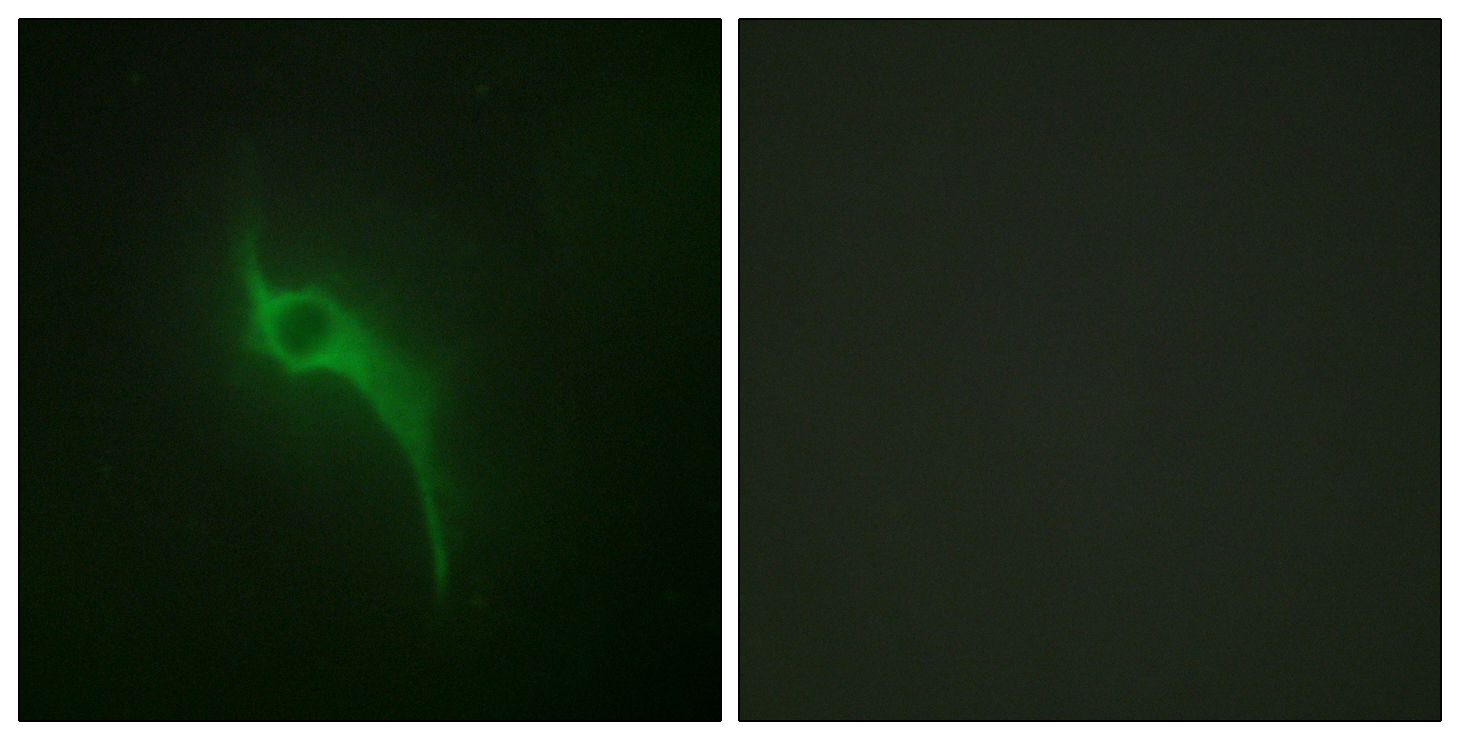
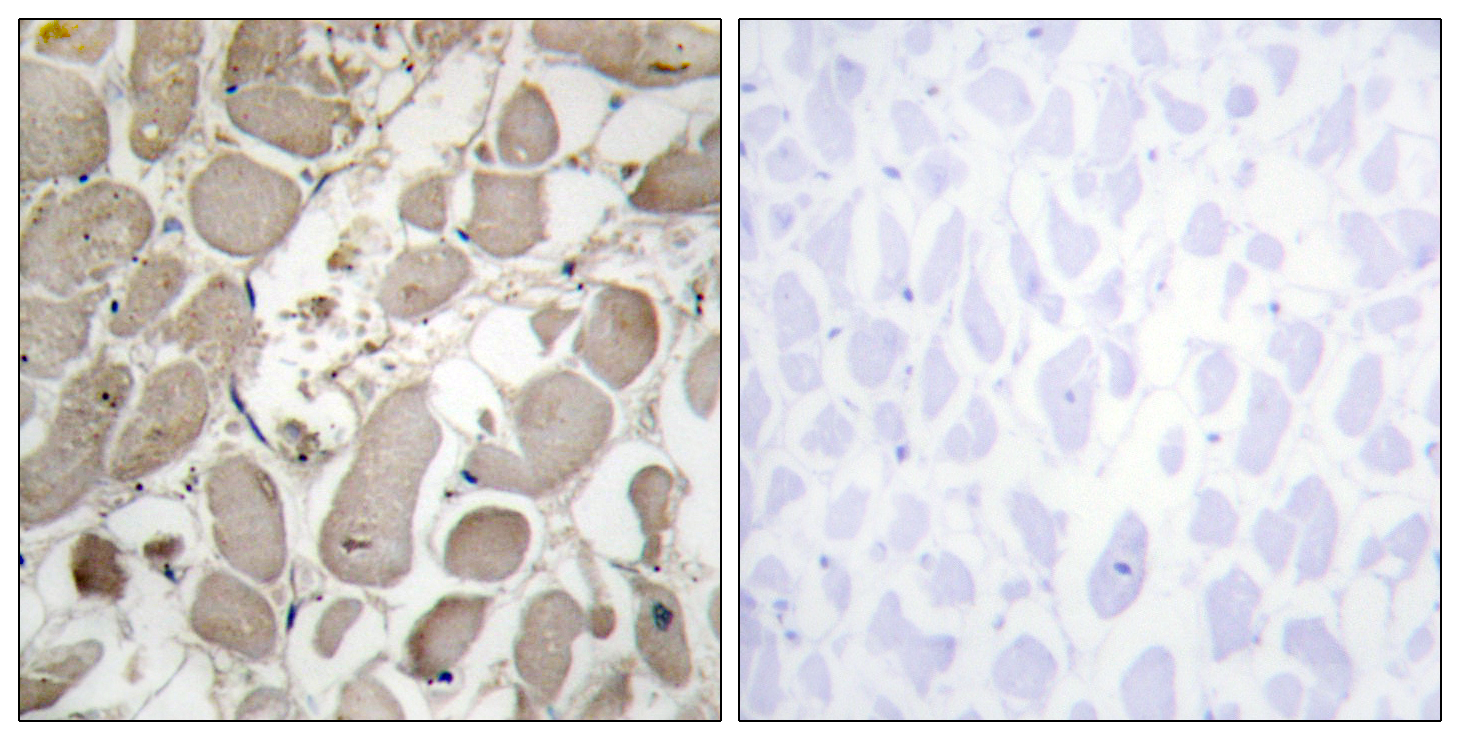
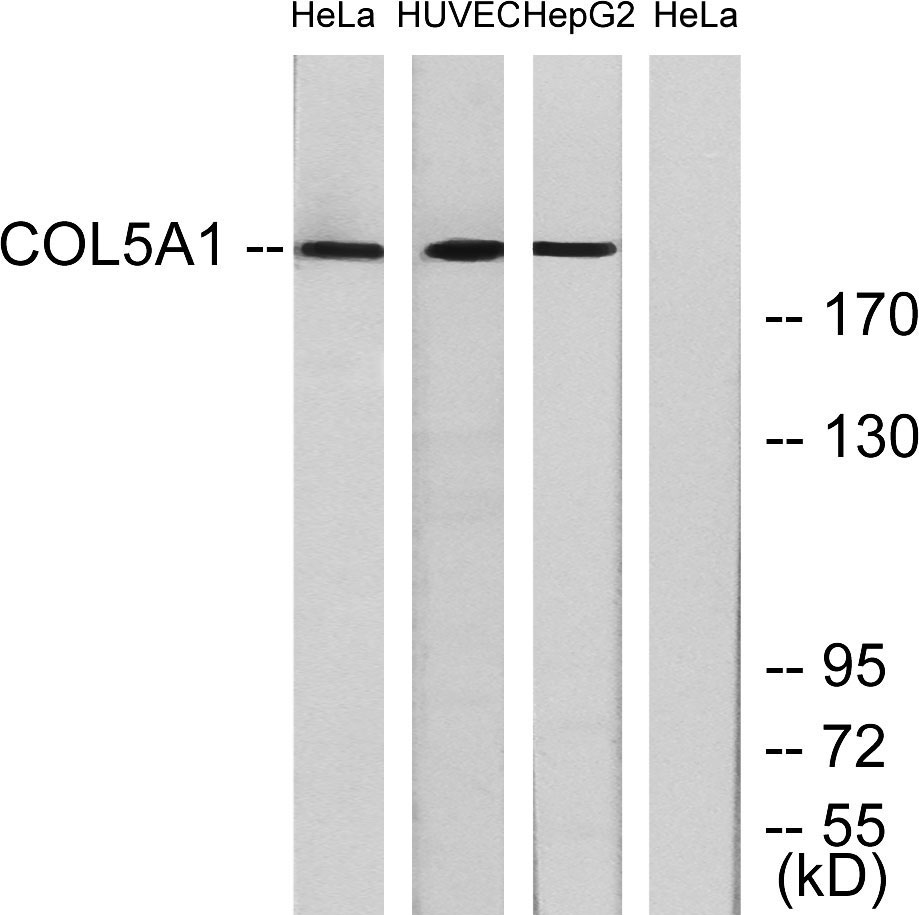
COL5A1 Rabbit Polyclonal Antibody
别名
COL5A1; Collagen alpha-1(V) chain
蛋白名称
Collagen alpha-1(V) chain
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=1289
Human Swissprot No.
P20908
Human Swissprot Link
http://www.uniprot.org/uniprotkb/P20908/entry
Mouse Swissprot No.
O88207
Mouse Swissprot Link
http://www.uniprot.org/uniprot/O88207
免疫原
The antiserum was produced against synthesized peptide derived from human Collagen V alpha1. AA range:301-350
特异性
COL5A1 Polyclonal Antibody detects endogenous levels of COL5A1 protein.
宿主
Polyclonal, Rabbit,IgG
背景介绍
This gene encodes an alpha chain for one of the low abundance fibrillar collagens. Fibrillar collagen molecules are trimers that can be composed of one or more types of alpha chains. Type V collagen is found in tissues containing type I collagen and appears to regulate the assembly of heterotypic fibers composed of both type I and type V collagen. This gene product is closely related to type XI collagen and it is possible that the collagen chains of types V and XI constitute a single collagen type with tissue-specific chain combinations. The encoded procollagen protein occurs commonly as the heterotrimer pro-alpha1(V)-pro-alpha1(V)-pro-alpha2(V). Mutations in this gene are associated with Ehlers-Danlos syndrome, types I and II. Alternative splicing of this gene results in multiple transcript variants. [provided by RefSeq, May 2013],
组织表达
Aorta endothelial cell,Chorioamniotic membrane,Eye,Placenta,
细胞定位
Secreted, extracellular space, extracellular matrix .
信号通路
Focal adhesion;ECM-receptor interaction;
功能
disease:Defects in COL5A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis or severe classic type Ehlers-Danlos syndrome. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome.,disease:Defects in COL5A1 are a cause of Ehlers-Danlos syndrome type 2 (EDS2) [MIM:130010]; also known as Ehlers-Danlos syndrome mitis or mild classic type Ehlers Danlos syndrome.,function:Type V collagen is a member of group I collagen (fibrillar forming collagen). It is a minor connective tissue component of nearly ubiquitous distribution. Type V collagen binds to DNA, heparan sulfate, thrombospondin, heparin, and insulin.,PTM:Prolines at the third position of the tripeptide repeating unit (G-X-Y) are hydroxylated in some or all of the chains.,PTM:Sulfated on 40% of tyrosines.,similarity:Belongs to the fibrillar collagen family.,similarity:Contains 1 laminin G-like domain.,similarity:Contains 1 TSP N-terminal (TSPN) domain.,subunit:Trimers of two alpha 1(V) and one alpha 2(V) chains in most tissues and trimers of one alpha 1(V), one alpha 2(V), and one alpha 3(V) chains in placenta. Interacts with CSPG4.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.