产品名称
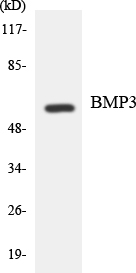
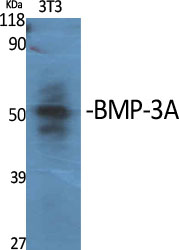
BMP-3A Rabbit Polyclonal Antibody
别名
BMP3; BMP3A; Bone morphogenetic protein 3; BMP-3; Bone morphogenetic protein 3A; BMP-3A; Osteogenin
蛋白名称
Bone morphogenetic protein 3
存储缓冲液
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N.
Human Gene Link
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&term=651
Human Swissprot No.
P12645
Human Swissprot Link
http://www.uniprot.org/uniprotkb/P12645/entry
Mouse Swissprot No.
Q8BHE5
Mouse Swissprot Link
http://www.uniprot.org/uniprot/Q8BHE5
免疫原
The antiserum was produced against synthesized peptide derived from human BMP3. AA range:281-330
特异性
BMP-3A Polyclonal Antibody detects endogenous levels of BMP-3A protein.
宿主
Polyclonal, Rabbit,IgG
背景介绍
This gene encodes a secreted ligand of the TGF-beta (transforming growth factor-beta) superfamily of proteins. Ligands of this family bind various TGF-beta receptors leading to recruitment and activation of SMAD family transcription factors that regulate gene expression. The encoded preproprotein is proteolytically processed to generate each subunit of the disulfide-linked homodimer. This protein suppresses osteoblast differentiation, and negatively regulates bone density, by modulating TGF-beta receptor availability to other ligands. [provided by RefSeq, Jul 2016],
组织表达
Expressed in adult and fetal cartilage.
功能
function:Negatively regulates bone density. Antagonizes the ability of certain osteogenic BMPs to induce osteoprogenitor differentitation and ossification.,induction:Highly expressed in fracture tissue, particularly in osteoblasts, osteoclasts and chondroblasts.,online information:Bone morphogenetic protein 3 entry,similarity:Belongs to the TGF-beta family.,subunit:Homodimer; disulfide-linked.,tissue specificity:Expressed in adult and fetal cartilage.,
纯化
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
.jpg)

