2021-12-10 阅读量:267
编者按
肌萎缩侧索硬化症(ALS)是一种致命的神经退行性疾病,由来自大脑和脊髓的运动神经元的缺失引起。该病导致四肢、躯干、胸部、腹部肌肉逐渐无力并萎缩,从而影响运动、交流、吞咽和呼吸功能,最终致死。在过去的近三十年里,ALS相关研究通过识别新的家族性突变基因,构建了ALS动物模型,研究疾病发生的分子机制,并最终开发出具有治疗前景的方案,并且有一部分治疗方案已经进入临床试验。结合基础和临床相关研究,小编在这里给大家推荐Kim G等发表在《Neuron》题为“ALS Genetics: Gains, Losses, and Implications for Future Therapies”的综述,该文围绕ALS的发病机制、相关基因治疗方案进行主要论述。
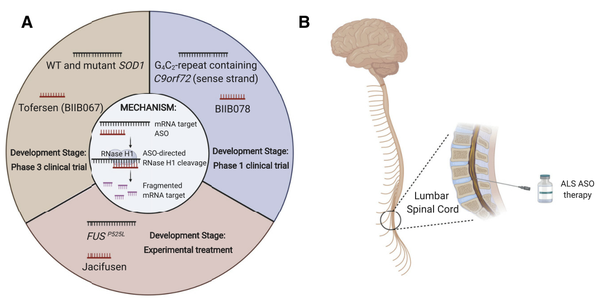
从发现第一个ALS致病基因至今的近30年时间里,ALS相关的治疗学研究已经步入临床治疗阶段。而在过去的十年中,反义寡核苷酸(ASOs)的开发和临床应用掀起基因治疗风暴。ASOs的思路非常简单,即如果突变的基因导致蛋白毒性而致病,那么就通过基因干预的方式来抑制毒性蛋白的基因转录,ASOs由此应运而生。目前通过靶向三种ALS致病基因(SOD1,C9ORF72和FUS)的ASOs正在进行临床研究。
大约有10%的ALS相关病例被认为是家族性ALS(fALS),其余的病例被认为是散发性ALS(sALS)。尽管fFALS不如sALS常见,但通过对家族性ALS相关的突变基因进行基因干预,对于理解ALS的发病机制有重大意义。目前为止,绝大多数的ALS突变基因的发现均是通过fALS获得,并认为突变基因通过获得功能(GOF,gain-of-function)相关机制,导致ALS的发生。目前认为GOF机制是ALS发病的关键因素,例如疾病的显性遗传模式、毒性蛋白质聚集体的产生,以及生物学过程的分子功能的变化。
1993年发现第一个ALS疾病基因超氧化物歧化酶1(SOD1),SOD1是通过GOF机制导致ALS。此外其他大多数ALS致病基因,如C90RF72、TARDBP、FUS和HNRNPA1,也可能通过GOF机制发挥作用。但是ALS的发病机制也可能是通过基因的丧失功能的(LOF,loss-of-function)突变,导致疾病的发生,如OPTN和TBK1中的突变。但是,即使被认为主要通过GOF机制的突变基因,如SOD1和C9ORF72,也表现出LOF特性,例如C9ORF72突变患者会出现免疫功能障碍。
一、SOD1
对导致ALS的不同SOD1突变体进行系统分析,发现SOD1的酶活性与ALS患者的发病与疾病进展之间存在相关性,并且有充分的证据支持SOD1的GOF机制导致ALS。例如,表达人类ALS相关变体SOD1G93A、SOD1G37R和SOD1G85R的转基因小鼠表现出明显的肌萎缩侧索硬化症样的表型,包括线粒体功能障碍、SOD1聚集、运动神经元死亡和运动能力障碍。并且无论内源性SOD1的存在与否,均不能改善SOD1G85R转基因小鼠的存活时间,表明SOD1通过GOF介导ALS表型。
另一方面,针对SOD1的LOF能明显改善运动系统功能异常。但是当将SOD1功能沉默作为ALS的潜在治疗方法时,必须考虑LOF对正常生理功能的潜在效应。目前靶向SOD1的ASOs方案初步的临床证据表明,SOD1的LOF对SOD1-ALS患者是安全的。
二、C90RF72
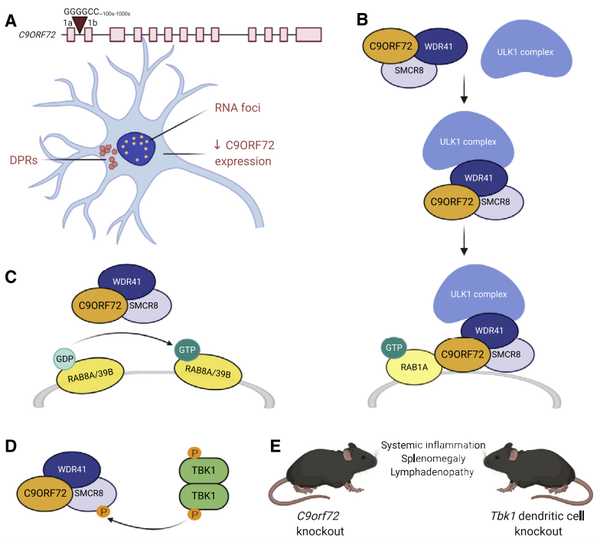
ALS最常见的遗传原因是由于9号染色体开放阅读框72基因(C9ORF72)出现GGGGCC(G4C2)六核苷酸重复扩增引起。在欧洲血统的人群中,这种突变是最常见的,约占ALS的34%,占家族性ALS的近四分之一。在健康人群C9ORF72中可能有2到25个G4C2重复,但ALS患者可出现成百上千个这样的重复。C9ORF72基因不仅在中枢神经系统中表达,在外周如淋巴细胞、骨髓、脾脏等也有表达,但其功能仍不完全清楚。C9ORF72非编码区的G4C2重复序列的扩张被认为至少通过以下三种主要机制中的一种引起ALS:(1)重复的RNA介导的细胞毒性,(2)二肽重复蛋白(DPR)介导的细胞毒性,(3)单倍体功能不足导致C9ORF72功能蛋白水平的降低。
研究发现C9ORF72基因敲除小鼠出现明显的免疫系统失调,表现为淋巴结肿大、脾肿大、自身免疫、细胞因子释放增加以及巨噬细胞和淋巴细胞在多个器官中存在大量浸润。此外,抑制干扰素基因刺激因子(STING)信号通路可以挽救在C9ORF72基因敲除小鼠中观察到的免疫缺陷。C9ORF72基因敲除小鼠和TBK1树突状细胞基因敲除小鼠具有相似的免疫系统的表型。研究发现C9ORF72与SMCR8和WDR41形成复合物,并通过依赖Rab1a的方式将ULK1复合物招募到噬菌体组装位点,从而在自噬启动过程中发挥作用。C9ORF72/SMCR8/WDR41复合体是参与自噬的Rab8a和Rab39b GTP酶的GDP/GTP交换因子,而TBK1与C9ORF72/SMCR8/WDR41复合物相互作用,可通过磷酸化SMCR8调节自噬。此外C9ORF72还可调控转录因子TFEB,TFEB也是自噬和溶酶体基因的主要调控因子。鉴于蛋白质质量控制缺陷导致的细胞质蛋白聚集体的形成是ALS和许多其他神经退行性疾病的关键病理标志,因此C9ORF72通过自噬影响这些蛋白质降解和清除与疾病的发生密切相关。
三、TDP-43
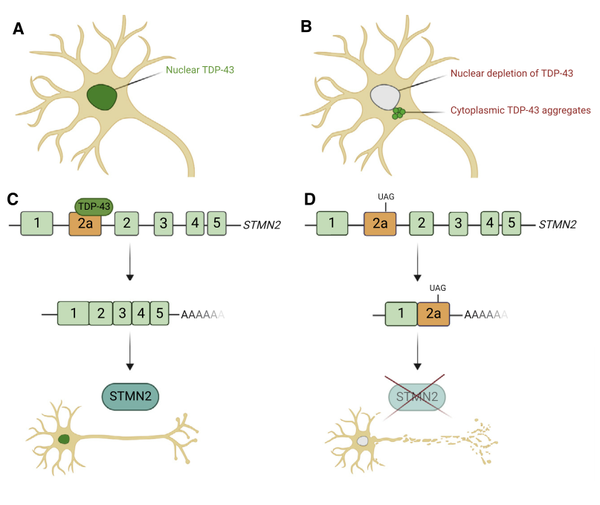
TDP-43被TARDBP基因所编码,是一种DNA/RNA结合蛋白,通常定位于细胞核。TDP-43调节RNA代谢的各个步骤,包括mRNA剪接、转运、翻译和microRNA合成。TDP-43是ALS中特征最明显的病理蛋白。在生理条件下,TDP-43主要定位于细胞核,并在RNA代谢中发挥重要作用。而在大多数ALS病例中,TDP-43在细胞核中被耗竭,并在细胞质中形成聚集体。因此TDP-43可能通过两方面参与ALS的病理过程。一是核内TDP-43正常功能丢失;二是TDP-43细胞质聚集体的形成通过细胞毒性的GOF参与ALS的发生。
TDP-43的剪接靶点stathmin-2(STMN2)有可能在ALS中发挥关键作用。研究发现下调TDP-43水平会导致数百个基因的表达发生变化,其中微管稳定调节因子STMN2是下调最严重的基因之一。在STMN2基因含有一个特殊的外显子(外显子2a),该外显子通常不转录在成熟的STMN2 mRNA中,但是当TDP-43功能受损时,外显子2a就会被整合到成熟的mRNA中。而这个外显子中含有一个终止密码子和一个多聚腺苷酸信号,可导致STMN2 mRNA翻译提前结束。虽然STMN2是一个潜在的有吸引力的ALS治疗靶点,但需要在更大规模的ALS患者中检测STMN2水平的变化情况。值得注意的是,STMN2中外显子2a在小鼠基因中并不保守,因此需要人源化基因的小鼠模型进行相关研究。
四、FUS/TLS
FUS/TLS(肉瘤融合/在脂肪肉瘤转运蛋白)突变可导致ALS。到目前为止,已经在ALS患者发现了50多种不同的FUS突变,其中许多突变干扰了核定位信号,并导致FUS错误定位到细胞质。在ALS患者死后的大脑尸检结果中,FUS阳性内含体存在于细胞质中,而较少出现在神经元和胶质细胞的细胞核。
ALS的FUS发病机制类似于TDP-43的发病机制,一是可能由于FUS聚集引起的毒性GOF;二是由于FUS胞浆错误定位和细胞核功能丧失引起的LOF。外源性表达突变的FUS或没有核定位信号的FUS的小鼠会表现出类似ALS的表型,支持了FUS的GOF假说。但是在斑马鱼中,敲除FUS基因同样会导致运动能力受损,运动神经元轴突投射缩短,以及神经肌肉接头功能出现障碍,提示FUS的LOF可能介导ALS。
五、OPEN和TBK1
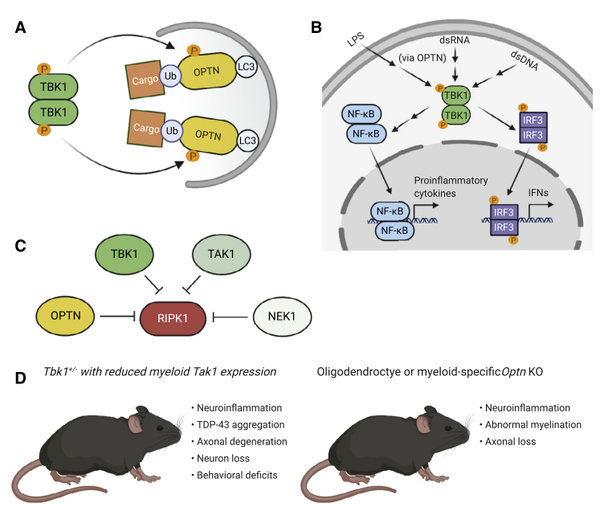
视神经蛋白基因(OPTN),也被称为FIP-2,首次在日本近亲婚姻的病例中发现OPTN突变导致ALS。OPTN突变包括:(1)第五外显子缺失,导致移码和终止密码子提前;(2)无义突变(p.Q398X)产生截短蛋白;(3)杂合错义突变(p.E478G),导致神经肌肉接头的OPTN表达降低,表明OPTN的LOF导致ALS。支持ALS的LOF的实验证据有,OPTN基因敲除导致斑马鱼胚胎中轴突运输动力学的改变和细胞死亡的增加,同样的,敲除N2a(小鼠脑神经瘤细胞)和小鼠视网膜神经节细胞(RGC)中的OPTN基因导致细胞死亡增加。
OPTN也受到TANK结合激酶1(TBK1)的调节,TBK1磷酸化OPTN,不仅能增加OPTN对泛素化底物的亲和力,还增加了其与LC3的结合清除损伤的蛋白。除了在自噬相关的途径发挥作用,OPTN和TBK1在先天免疫系统中也有共同的作用。
六、当前临床进展
在SOD1-ALS动物模型中使用ASOs成功靶向沉默SOD1的mRNA,基于此设计的ASOs治疗剂tofersen(BIIB067)正式进入临床实验。在一项针对50名SOD1-ALS患者中,接受100毫克tofersen的患者脑脊液中SOD1浓度下降了36%,而对照组仅下降了3%。显示出ASOs在ALS广阔的治疗前景。而在针对C9ORF72的ASOs靶向沉默技术在临床前研究也显示出良好的疗效。在针对人类C9ORF72-ALS患者的ASOs治疗学研究中,ASOs BIIB078正在接受一期临床的安全性评价研究。
七、ALS的多靶点治疗方案
虽然许多ALS基因的突变已经证明通过GOF机制介导毒性反应,但这种效应通常与LOF效应有内在联系,LOF效应也可能导致疾病的发生和发展。因此,同时针对GOF和LOF机制的多靶点治疗方法对于治疗大多数ALS患者显得尤为必要。例如,C9ORF72中的G4C2重复扩增导致RNA聚集、DPRs和C9ORF72水平降低。虽然G4C2重复的靶向ASOs的目的是减轻RNA和DPR介导的毒性反应,但该方案并不能增加C9ORF72水平。因此,靶向包含正义和反义G4C2重复序列的RNA,并提高C9ORF72水平的联合疗法,可能在改善这种突变引起的LOF和毒性GOF效应方面更有效。
八、展望
在不到三十年的研究历程中,ALS相关研究已经步入临床治疗阶段。为单基因突变患者提供个性化的ASOs治疗策略有可能成为ALS的常规治疗方案,但是在治疗ALS的过程中,需要同时考虑功能丧失(LOF)和功能获得(GOF),因为一些LOF效应可以显着改变疾病的相关表型。总而言之,本综述系统阐述了ALS相关功能丧失和功能获得方面的研究进展,而针对GOF和LOF的结合治疗方案,有可能成为治愈ALS的有效策略。
图片来源:Neuron
原文链接:https://doi.org/10.1016/j.neuron.2020.08.022
扫码阅读原文

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK