2025-11-24 阅读量:686
肥胖发生率上升正推动全球非酒精性脂肪性肝病(NAFLD)流行。NAFLD包含两个阶段:以脂质过度蓄积为特征的单纯性脂肪肝(NAFLD早期)和标志疾病进展的非酒精性脂肪性肝炎(NASH)。脂质代谢失衡是NAFLD的主要病因。肝脏脂质调控涉及脂肪酸摄取、从头脂质生成(DNL)和脂质氧化等复杂过程。DNL是将碳水化合物转化为脂肪酸的严格调控过程,依赖胰岛素信号通路及关键转录因子如固醇调节元件结合蛋白1c(SREBP-1c)和肝X受体(LXRs)。SREBP-1c上调DNL通路中脂肪酸合酶(Fasn)和硬脂酰辅酶A去饱和酶1(Scd1)的基因表达。在肥胖等条件下,失控的DNL导致甘油三酯(TG)过度蓄积,因此调控肝脏脂质生成成为NAFLD治疗的潜在策略。
白细胞介素22(IL-22)是由固有淋巴细胞分泌的蛋白,具有组织修复功能,并通过防止肝细胞凋亡、调节脂质代谢和增强胰岛素敏感性来保护肝脏免受NAFLD侵害。这些作用涉及信号转导和转录激活因子3(STAT3)和蛋白激酶B(Akt)信号通路。IL-22通过与由IL-22受体α1亚基(IL22RA1)和IL-10受体β亚基组成的受体复合物结合而发挥信号作用。IL22RA1作为IL-22的功能性受体,特异性表达于肝脏、胰腺和肠道等代谢组织。此外,IL22RA1还与IL20R2结合形成IL-20和IL-24的受体。值得注意的是,仅IL-22能激活肝细胞中的STAT3,而IL-20和IL-24对代谢紊乱的影响微乎其微。由于IL-22/IL22RA1通路具有组织保护和代谢调节作用,且不影响免疫系统,靶向该通路为NAFLD治疗提供了新希望。然而,IL22RA1在肝细胞中的精确功能及其在NAFLD中的作用仍需进一步探索。
氧化甾醇是代谢调控的关键因子,具有治疗潜力。它们是由胆固醇通过特定反应产生的生物活性脂质,影响免疫、炎症过程和代谢综合征。胆固醇稳态破坏导致的氧化甾醇失衡会损害代谢稳态。如,17-羟孕酮(胆固醇转化为皮质醇过程中形成的类固醇中间体)的全身给药或肝脏蓄积会导致小鼠高血糖和胰岛素抵抗(IR)。相反,25-羟基胆固醇水平升高可减轻肝脂肪变性。尽管氧化甾醇在代谢疾病中至关重要,但内源性氧化甾醇与肝脏脂质代谢之间的具体联系尚未完全明确。
近期,上海交通大学医学院附属第六人民医院胡承研究员/陆炎研究员团队在肝病学领域顶刊Hepatology(IF=12.9)发表以“Hepatic IL22RA1 deficiency promotes hepatic steatosis by modulating oxysterol in the liver”为题的研究论文。作者发现,在棕榈酸(PA)处理的小鼠原代肝细胞(MPHs)、NAFLD模型小鼠及患者的肝脏组织中,IL22RA1表达均下调。肝细胞特异性Il22ra1基因敲除(HKO)小鼠在高脂饲料(HFD)和高果糖高胆固醇(HFHC)饲料喂养下,均表现出加重的肝脂肪变性。其机制为:肝细胞中IL22RA1缺失通过氧化甾醇——3β-羟基-5-胆甾烯酸(3βHCA)介导的脂质生成加剧肝脂肪变性,该过程涉及激活转录因子3(ATF3)/氧化甾醇7α-羟化酶(CYP7B1)信号轴。此外,HFD喂养条件下,HKO小鼠肝脏中恢复CYP7B1表达或沉默ATF3,均可降低肝脏3βHCA水平及脂质含量。这些研究结果揭示了3βHCA的促脂质生成作用,并彰显肝细胞IL22RA1在NAFLD进展中的调控作用。

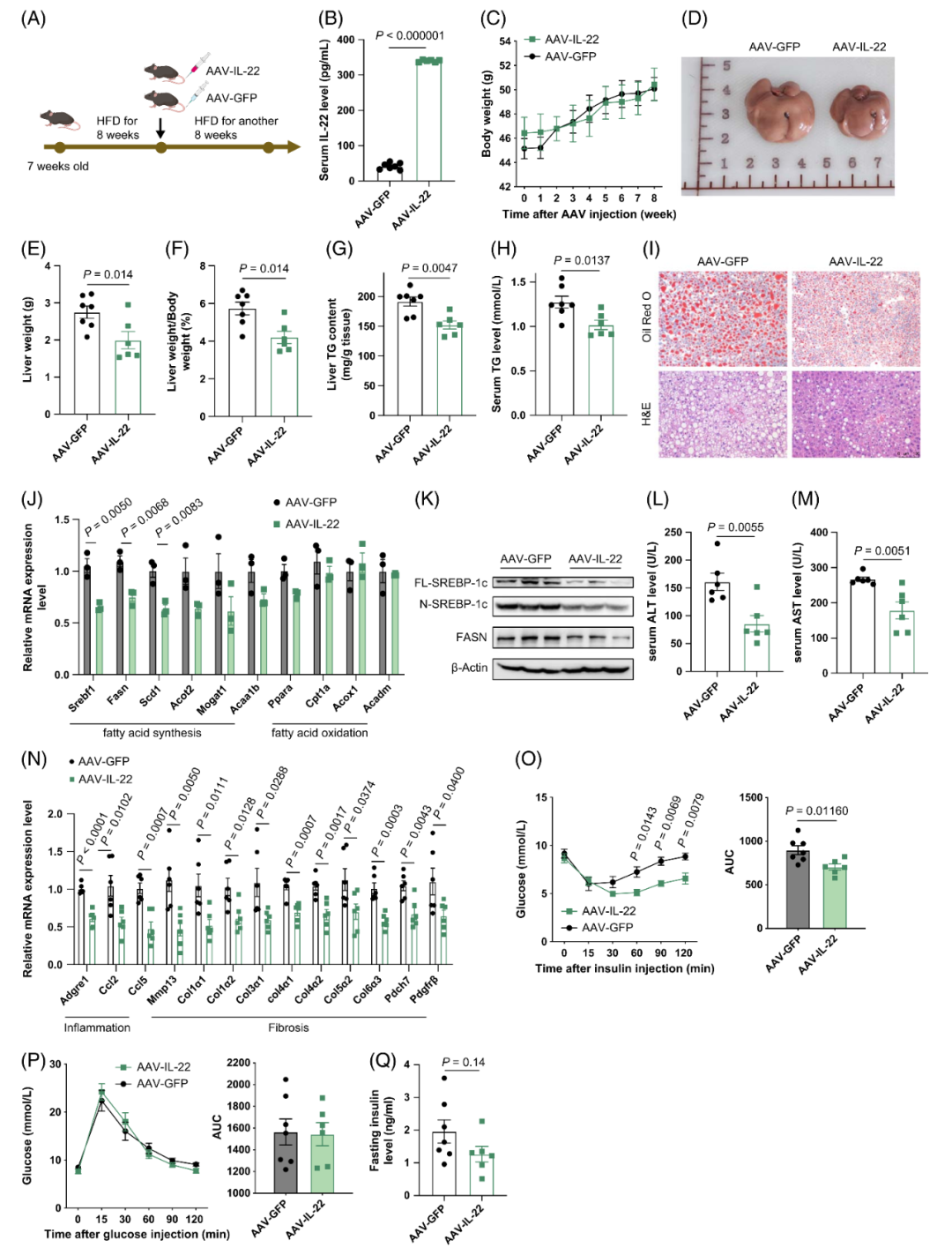
1.IL-22过表达改善HFD诱导的肝脂肪变性
为评估腺相关病毒(AAV)介导的IL-22过表达对肝脂肪变性的影响,研究人员将7周龄C57BL/6J小鼠HFD喂养8周后,通过尾静脉注射AAV-IL-22或AAV-GFP。酶联免疫吸附试验(ELISA)验证了AAV-IL-22组小鼠血清IL-22水平显著升高。尽管两组小鼠体重无显著差异,但IL-22过表达组小鼠的肝重、肝重/体重比均下降,且肝脂肪变性程度减轻——表现为肝脏TG水平低于对照组。肝组织切片油红O染色和苏木精-伊红(H&E)染色进一步证实,AAV-IL-22组小鼠肝脏脂质蓄积减少。实时荧光定量PCR(qPCR)和蛋白质免疫印迹(Western blot)结果显示,IL-22过表达显著下调生脂基因的表达[全长无活性SREBP-1c(FL-SREBP-1c)、其N端活性片段(N-SREBP-1c)及脂肪酸合酶(FASN)的蛋白表达水平均显著下降];然而,两组小鼠脂肪酸氧化相关基因的表达无显著差异。此外,HFD喂养的AAV-IL-22组小鼠,其肝损伤标志物[血清丙氨酸转氨酶(ALT)、天门冬氨酸转氨酶(AST)]及血清TG水平均显著降低。IL-22过表达还与炎症和纤维化相关基因的表达下调相关。胰岛素耐量试验(ITT,腹腔注射胰岛素)显示,与对照组相比,AAV-IL-22组小鼠胰岛素敏感性显著增强,但两组小鼠的空腹血清胰岛素水平和糖耐量无显著差异。上述数据表明,IL-22可缓解HFD诱导的NAFLD进展。
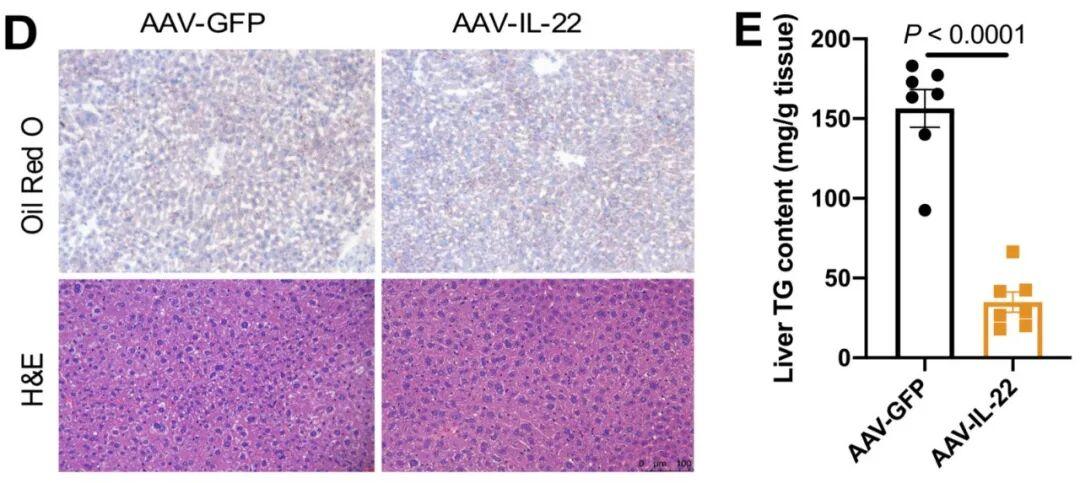
类似地,在标准饲料(CD)喂养条件下,AAV-IL-22组小鼠的肝脏TG水平仍低于对照组。肝组织切片油红O染色和H&E染色证实了这一降低趋势。这些结果表明,在模拟生理状态的CD喂养条件下,IL-22同样可降低小鼠肝脏TG含量。
图1.IL-22过表达对HFD诱导的肝脂肪变性具有保护作用
图2.IL-22过表达同样可减少CD喂养小鼠的肝脏脂肪蓄积
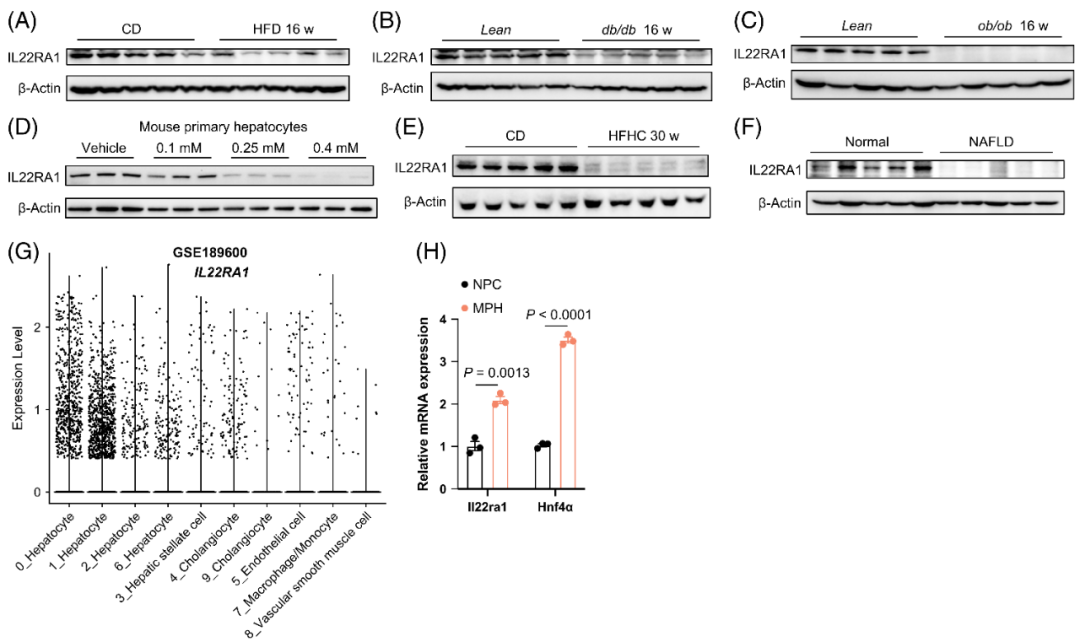
2.脂肪肝组织及PA处理MPHs中IL22RA1表达降低
为探究IL22RA1的内源性作用,研究者检测了其在3种NAFLD小鼠模型中的表达。肝脏样本的Western blot分析显示,与正常对照组相比,HFD(16 w)小鼠、db/db小鼠(瘦素受体基因突变纯合子)和ob/ob小鼠(瘦素基因突变纯合子)的肝脏IL22RA1蛋白水平均显著下调。PA处理MPHs的实验结果显示,随着PA浓度升高,IL22RA1表达逐渐降低。此外,与健康个体相比,HFHC饲料(30 w)诱导的NASH小鼠及NAFLD患者的肝脏IL22RA1蛋白水平均降低。基于公共单核RNA测序(snRNA-seq)数据(GSE189600)分析显示,在人类肝脏样本中,与肝脏非实质细胞(NPC)相比,肝细胞中IL22RA1 mRNA表达显著富集;这一结果通过MPHs的qPCR分析得到验证。
图3.IL22RA1在脂肪肝组织及PA处理MPHs中表达下调
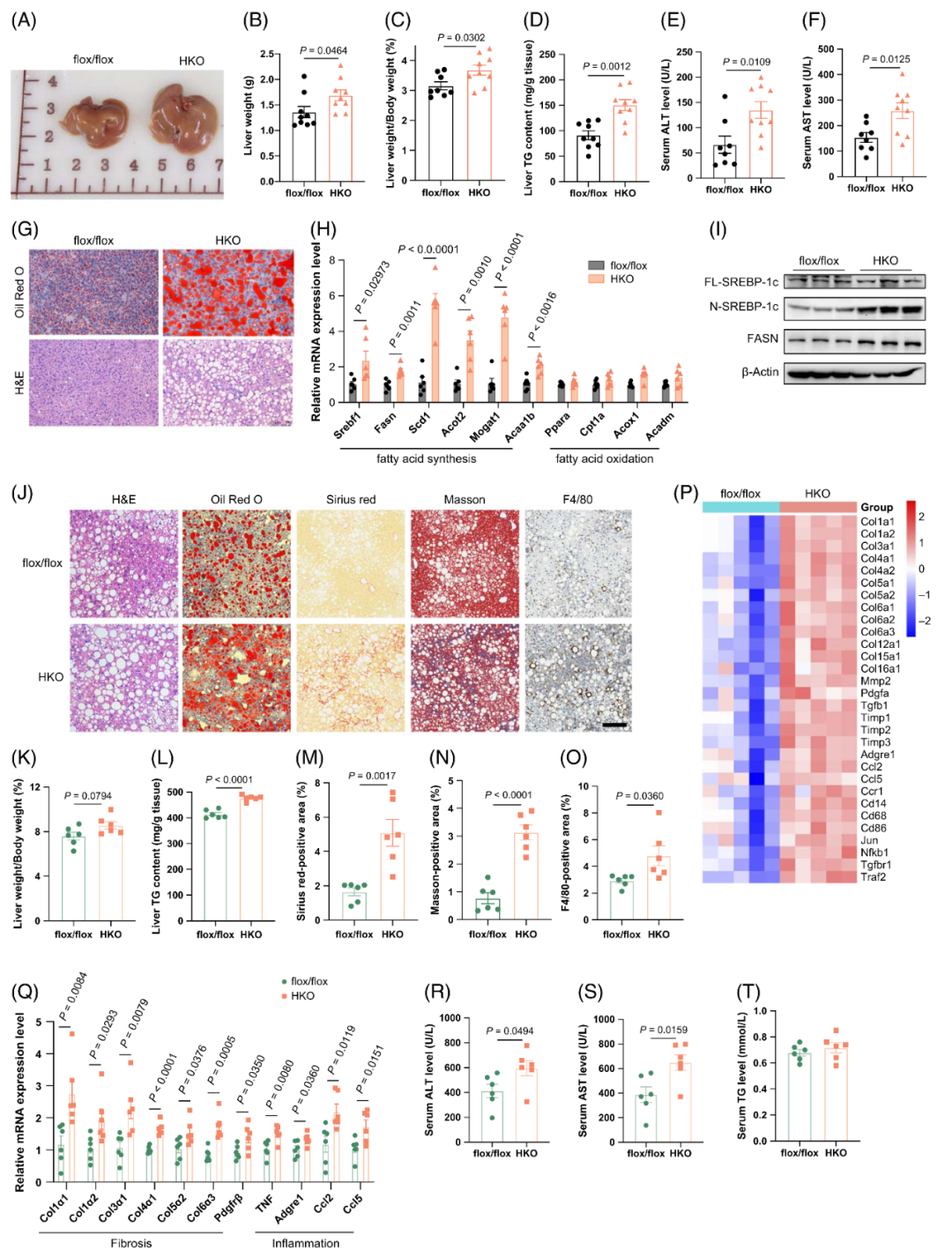
3.肝细胞特异性IL22RA1缺失加剧HFD诱导的肝脂肪变性及HFHC饲料诱导的NASH
为研究IL22RA1对肝脏脂质代谢的影响,作者对肝细胞特异性IL22RA1敲除(HKO)小鼠进行了分析。肝脏组织的qPCR、Western blot及免疫荧光染色结果证实,HKO小鼠肝脏中IL22RA1被成功敲除,且不影响其他组织的IL22RA1表达。HKO小鼠来源的原代肝细胞经IL-22刺激后,磷酸化STAT3(p-STAT3)水平未升高,表明IL22RA1敲除有效。
尽管CD喂养的HKO小鼠与同窝对照(flox/flox)小鼠的肝重、肝重/体重比、血清TG水平及体重无显著差异,但HKO小鼠肝脂肪变性程度呈加重趋势——表现为肝脏TG含量略高,脂质蓄积增加。此外,与对照组相比,HKO小鼠血清ALT和AST水平升高,糖耐量和胰岛素耐量略有受损,提示即使在CD喂养条件下,肝细胞IL22RA1缺失也会促进肝脏TG蓄积。
HFD喂养条件下,HKO小鼠的肝重、肝重/体重比及肝脏TG含量均显著高于flox/flox小鼠。油红O染色和H&E染色证实,HKO小鼠肝脏脂质蓄积显著增加。然而,两组小鼠的血清TG水平无显著差异。HKO小鼠肝脏中生脂基因表达显著上调,而脂肪酸β-氧化相关基因表达与对照组无显著差异,这与两组小鼠代谢率无明显变化的结果一致。与肝脂肪变性加重相对应,HKO小鼠血清ALT和AST水平显著升高,提示肝损伤加剧。此外,RNA-seq和qPCR结果显示,与对照组相比,HKO小鼠肝脏中炎症和纤维化相关基因表达显著上调。ITT和糖耐量试验(GTT,腹腔注射葡萄糖)结果显示,IL22RA1缺失加剧了IR和糖耐量异常。IR增强导致HKO小鼠空腹血清胰岛素水平高于对照组。综上,这些结果表明,肝细胞IL22RA1缺失会加剧HFD诱导的肝脂肪变性,并破坏血糖稳态。
研究人员进一步探究了肝细胞IL22RA1在HFHC饲料诱导的NASH中的作用。HFHC饲料喂养后,HKO小鼠与对照组体重无显著差异,但肝重、肝重/体重比略高。H&E染色、油红O染色及肝脏TG含量检测结果显示,HKO小鼠肝脂肪变性程度加重。天狼猩红(Sirius red)染色、Masson染色结果显示,HKO小鼠肝脏胶原沉积增加;同时,其肝脏纤维化相关基因mRNA表达水平显著高于flox/flox小鼠,提示肝纤维化加剧。肝组织切片F4/80免疫组化染色及炎症相关基因mRNA表达检测结果显示,HKO小鼠肝脏炎症反应较对照组更为明显。与flox/flox小鼠相比,HKO小鼠血清ALT和AST水平显著升高,而血清TG水平无显著变化。这些结果表明,肝细胞IL22RA1缺失会促进NASH的发生发展。
图4.肝细胞特异性IL22RA1缺失加重HFD诱导的肝脂肪变性及HFHC饲料诱导的NASH
4.IL-22过表达可诱导肝脏CYP7B1表达,而肝脏IL22RA1缺陷则下调其表达
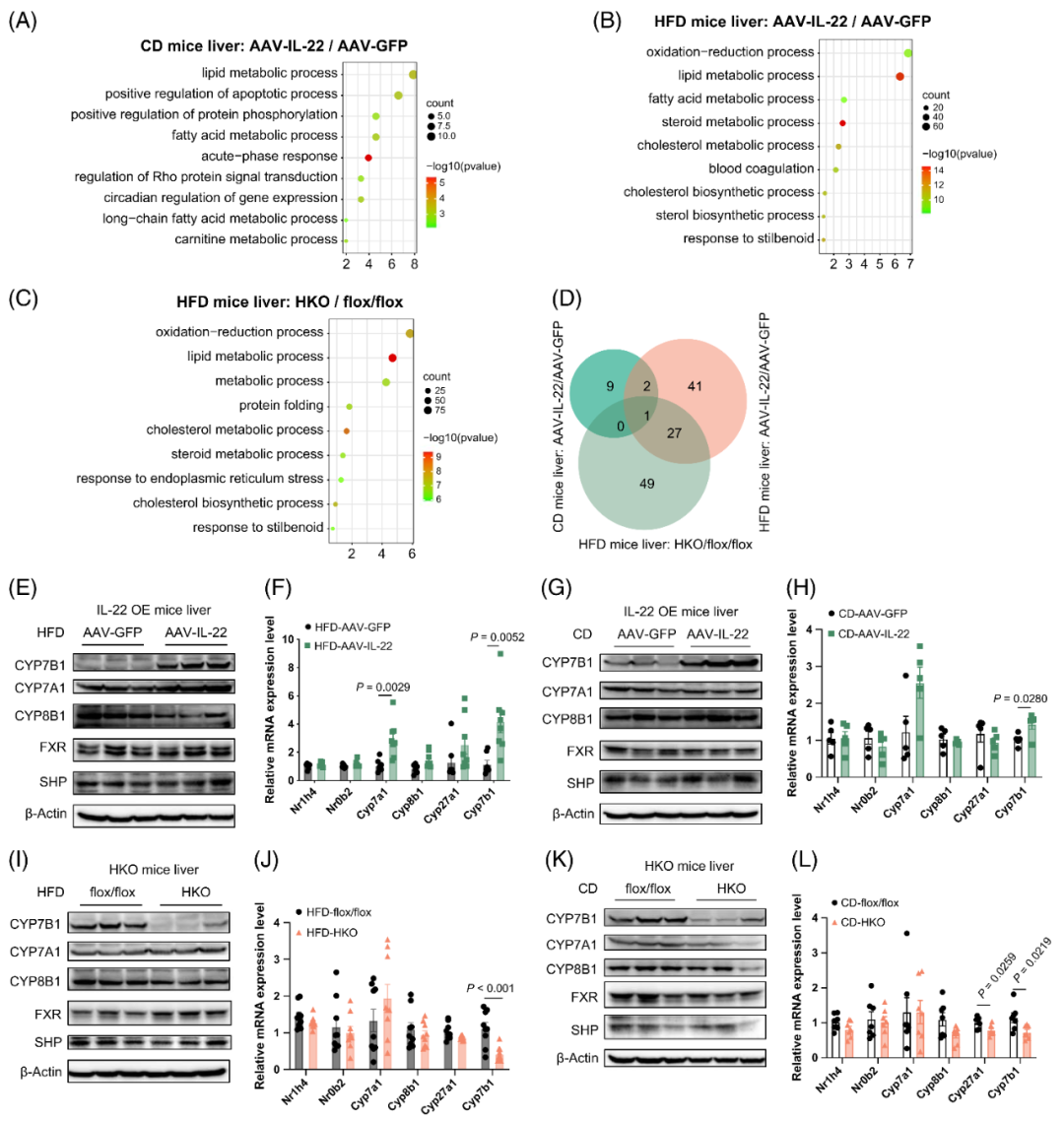
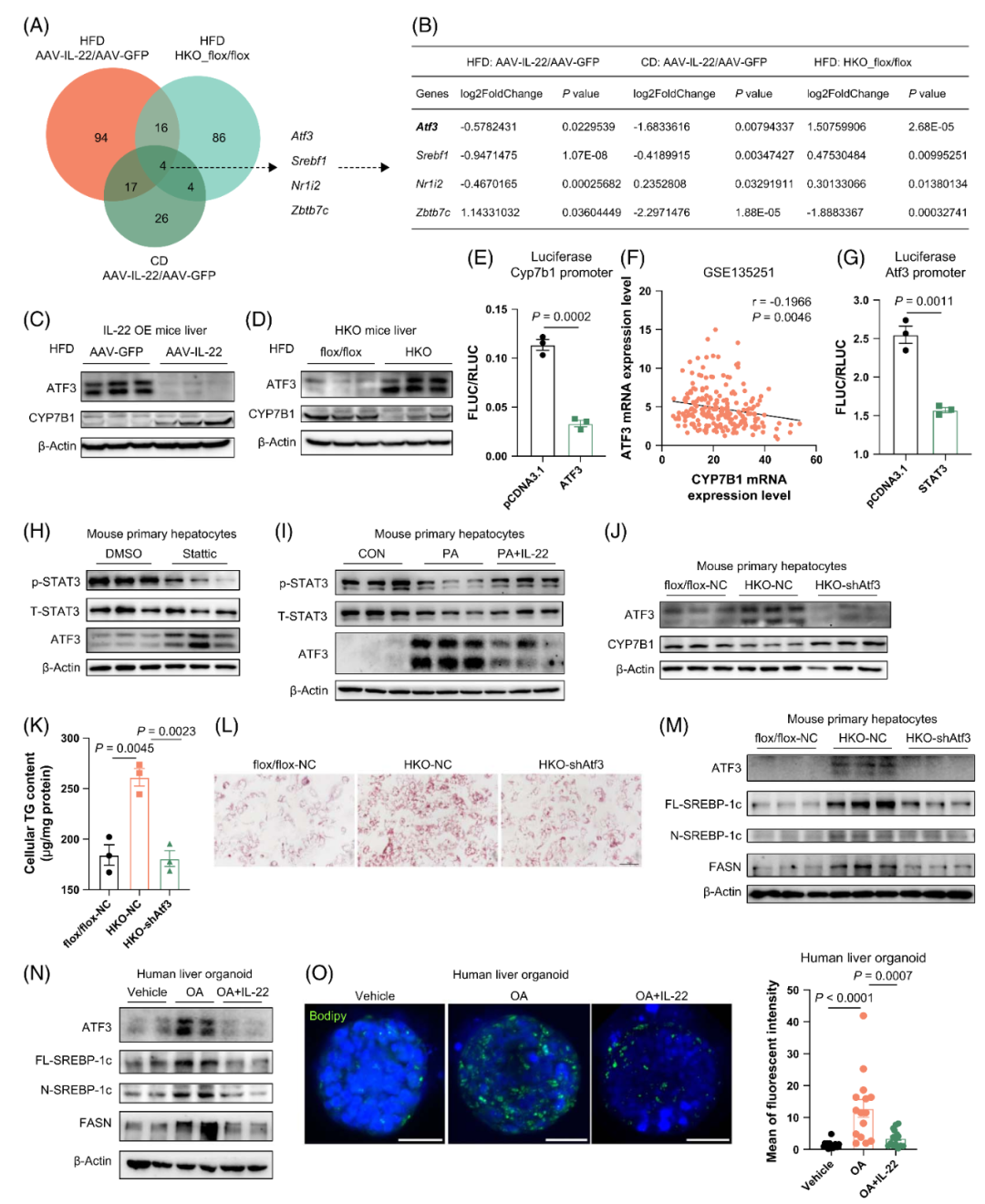
已证实IL-22过表达可改善肝脂肪变性,而肝细胞特异性IL22RA1缺陷会加重该病变,在此基础上,作者深入探究了IL-22/IL22RA1信号通路调控肝脏脂质代谢的分子机制。为此,对多种动物模型的肝脏组织进行了RNA-seq分析,包括:CD或HFD喂养的AAV-IL-22小鼠与AAV-GFP对照小鼠,以及HFD喂养的HKO小鼠与flox/flox对照小鼠。对以下三组样本的差异表达基因(DEGs)进行了基因本体(GO)分析:CD喂养的AAV-IL-22组与AAV-GFP组、HFD喂养的AAV-IL-22组与AAV-GFP组、HFD喂养的HKO组与flox/flox组。结果显示,在三组样本中,脂质代谢过程均为富集程度最高的生物学过程。研究人员在这三组中分别鉴定出12个、71个和77个与脂质代谢相关的DEGs。值得注意的是,Cyp7b1(编码氧化甾醇7α-羟化酶,该酶是胆汁酸合成替代途径中的关键酶)在三组中均为共同DEG。
随后,研究者在不同动物模型中验证了CYP7B1的表达水平。结果显示,无论是CD还是HFD喂养的AAV-IL-22小鼠,其肝脏中CYP7B1的蛋白和mRNA水平均显著升高,与RNA-seq结果一致。然而,其他胆汁酸合成相关酶的编码基因(如Cyp8b1和Cyp27a1)表达基本未受影响,仅HFD喂养IL-22过表达小鼠肝脏中Cyp7a1 mRNA水平显著升高。相反,与flox/flox对照小鼠相比,无论是CD还是HFD喂养的HKO小鼠,其肝脏CYP7B1表达均显著降低。flox/flox组与HKO组中Cyp7a1、Cyp8b1的表达基本无明显变化,仅CD喂养HKO小鼠肝脏中Cyp27a1 mRNA水平显著下降。此外,胆汁酸合成的关键调控因子——法尼醇X受体(FXR,由Nr1h4编码)和小异源二聚体伴侣(SHP,由Nr0b2编码)的表达水平,在IL-22过表达小鼠和IL22RA1缺陷小鼠的肝脏中与对照组相比均无显著差异。这些数据表明,CYP7B1可能是IL-22信号通路介导肝脏脂质代谢调控的关键靶基因。
图5.IL-22过表达可诱导肝脏中CYP7B1的表达,而IL22RA1缺陷则会降低其表达水平
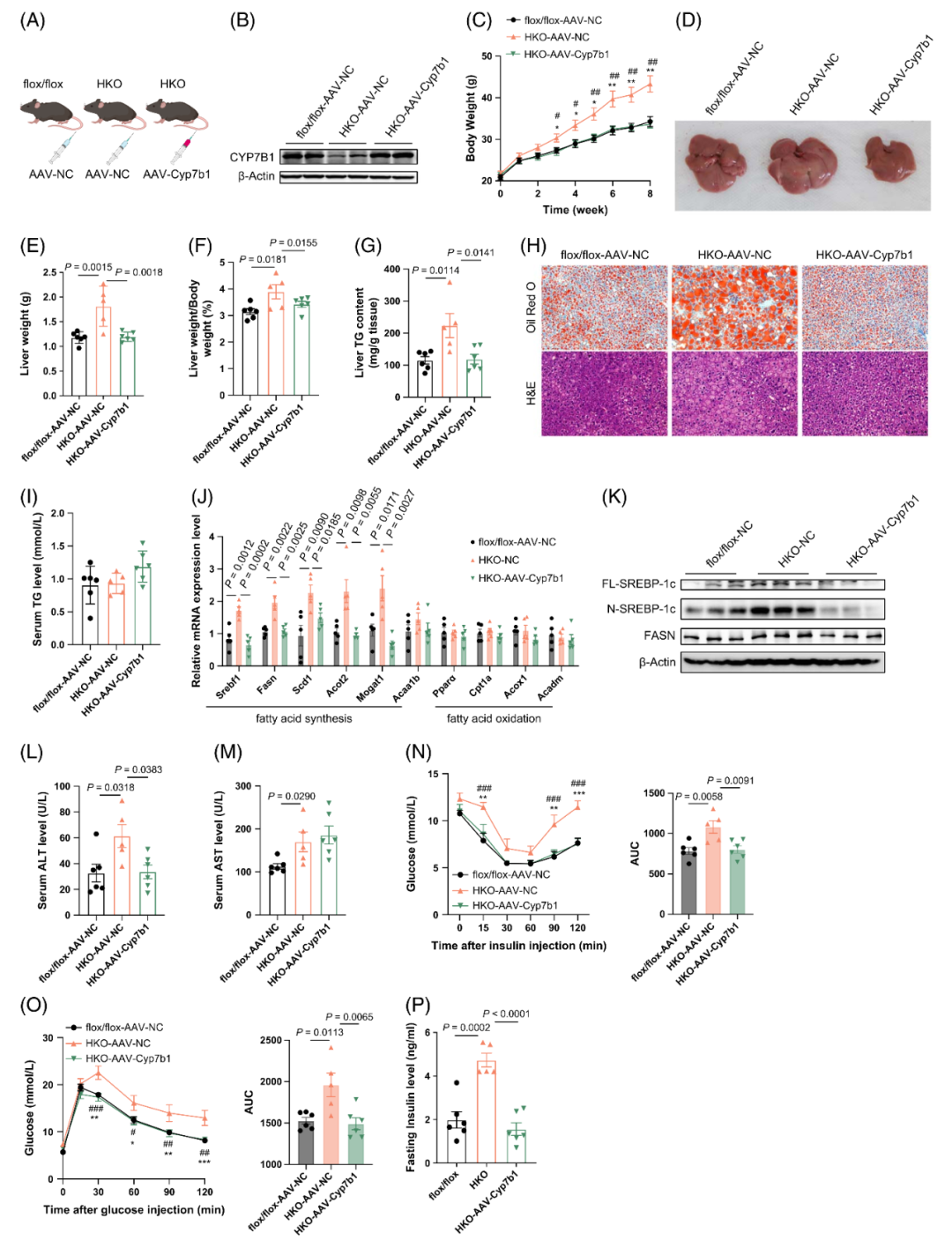
5.补充CYP7B1可逆转HKO小鼠中HFD诱导的肝脂肪变性
鉴于HKO小鼠肝脏CYP7B1水平降低,研究人员进一步探究补充CYP7B1是否能缓解该模型小鼠中HFD诱导的肝脂肪变性加重。通过AAV8病毒载体(分别携带GFP基因或小鼠CYP7B1基因)感染小鼠,以增强肝脏CYP7B1表达。结果显示,与flox/flox-AAV-NC(阴性对照)组小鼠相比,HKO-AAV-NC组小鼠肝脏CYP7B1表达显著降低,而HKO-AAV-Cyp7b1组小鼠肝脏CYP7B1呈过表达状态。
在HFD喂养条件下,HKO-AAV-Cyp7b1组小鼠体重增长减缓,且在HFD喂养4周后与HKO-AAV-NC组小鼠的体重差异显著,该差异在8周的观察期内持续扩大。两组小鼠肝脏的大体形态(大小和颜色)变化明显,其中HKO-AAV-Cyp7b1组小鼠的肝重、肝脏TG含量及肝重/体重比均显著降低。肝组织切片的油红O染色和HE染色结果证实,HKO-AAV-Cyp7b1组小鼠肝脏脂滴形成显著减少,这与该组小鼠肝脏生脂基因表达受抑制密切相关(相较于HKO-AAV-NC组)。
此外,CYP7B1过表达后,HKO-AAV-Cyp7b1组小鼠的血清ALT水平低于HKO-AAV-NC组,同时胰岛素敏感性和糖耐量显著改善,血清胰岛素水平降低;而血清TG和AST水平无明显变化。这些数据证实,CYP7B1过表达可逆转HKO小鼠的肝脂肪变性,同时提升其胰岛素敏感性和糖耐量。
图6.补充CYP7B1可逆转HKO小鼠中HFD诱导的肝脂肪变性
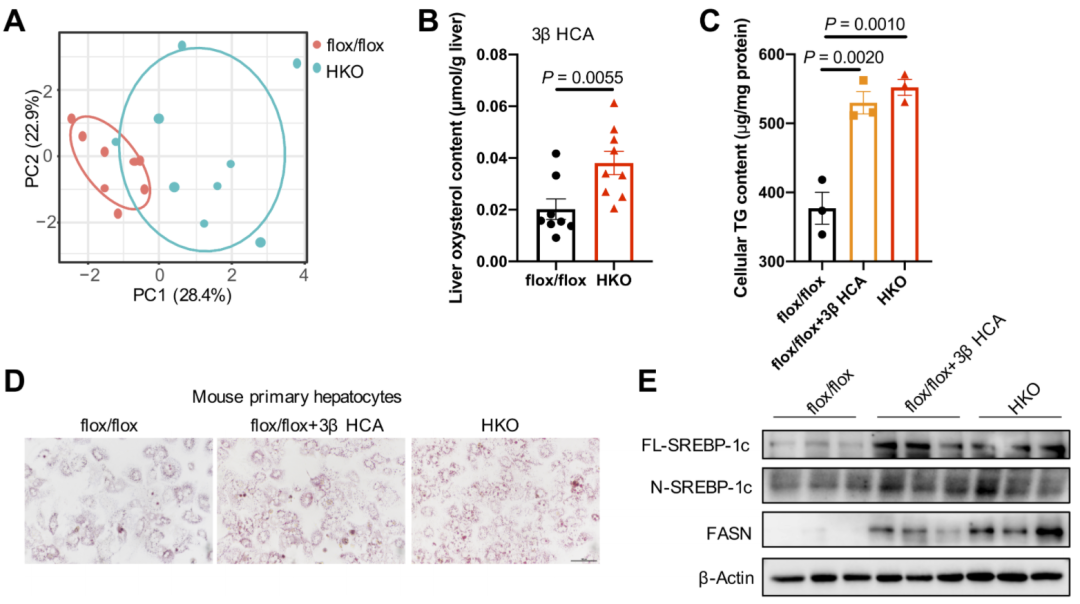
6.CYP7B1缺失导致的3β-羟基-5-胆甾烯酸(3βHCA)蓄积,会在MPHs及人类肝脏类器官中加速脂肪生成
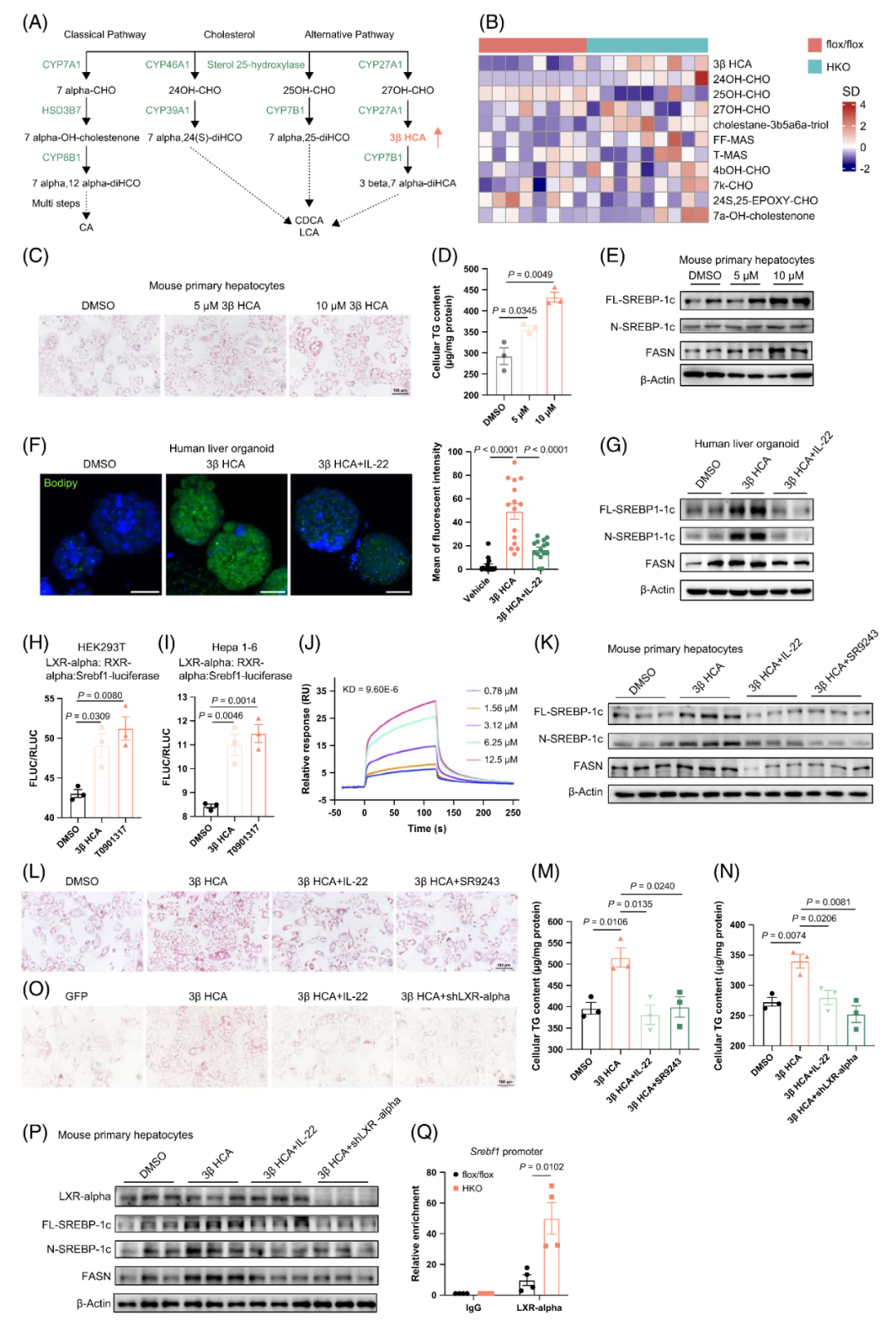
CYP7B1是胆固醇代谢与氧化甾醇生成过程中的关键酶。本研究探讨了其在HKO小鼠肝脂肪变性中的作用。对flox/flox小鼠(对照小鼠)和HKO小鼠肝组织的氧化甾醇分析显示,主成分分析(PCA)可将两组样本明确区分。在HKO小鼠肝脏中,3βHCA(胆固醇代谢过程中由固醇27-羟化酶CYP27A1催化27-羟基胆固醇转化生成)是所有检测的氧化甾醇中,与flox/flox小鼠相比升高最为显著的一种。CYP7B1可进一步将3βHCA转化为3β,7α-二羟-5-胆甾烯酸。用3βHCA处理MPHs后,细胞TG水平呈非剂量依赖性升高,油红O染色结果也证实了这一现象,其机制与生脂基因表达上调有关。类似地,人类肝脏类器官(HLOs)暴露于3βHCA后脂肪生成增强,Bodipy荧光染料(特异性标记并可视化细胞或组织中的脂质)染色及Western blot结果均验证了该效应。而IL-22处理可显著减弱3βHCA诱导的HLOs脂质合成。重要的是,用3βHCA处理flox/flox小鼠来源的原代肝细胞后,其细胞脂质沉积水平与HKO小鼠来源的原代肝细胞相当。
胆固醇代谢通路可将胆固醇转化为氧化甾醇,后者进一步生成胆汁酸。胆汁酸对脂质、葡萄糖代谢等多条代谢通路具有重要调控作用。为探究胆汁酸在脂质代谢调控中的作用,本研究分析了肝脏IL22RA1缺陷小鼠及IL-22过表达小鼠的血清胆汁酸水平。结果显示,HFD喂养的flox/flox小鼠与HKO小鼠相比,其血清中初级胆汁酸组成、总胆汁酸水平、初级与次级胆汁酸相对丰度、结合型与非结合型胆汁酸相对丰度,以及12-羟基(12OH)与非12-羟基胆汁酸相对丰度均无显著差异。此外,IL-22过表达对CD或HFD喂养小鼠的血清初级胆汁酸组成及水平影响极小。上述结果表明,肝脏IL22RA1缺陷小鼠出现的有害表型,可能与肝脏氧化甾醇(尤其是3βHCA)水平升高相关,而非胆汁酸的作用。
肝X受体(LXRs),尤其是LXR-α(编码基因Nr1h3),是氧化甾醇的关键作用靶点。为探究3βHCA是否通过结合LXR-α调控脂肪酸合成,本研究通过双荧光素酶报告基因实验评估了3βHCA对小鼠固醇调节元件结合转录因子1(Srebf1)基因启动子的转录激活能力。研究表明,LXR-α可与视黄醇X受体α(RXR-α)相互作用形成异源二聚体,该二聚体结合LXR反应元件(LXRE)后调控基因表达。研究人员向人胚肾293T(HEK293T)细胞及小鼠肝癌细胞(Hepa 1-6)中分别转入LXR-α、RXR-α表达质粒及Srebf1(LXR-α的靶基因)报告基因质粒后,结果显示3βHCA可在两种细胞中均诱导LXR-α的转录激活,其效应与合成型LXR-α激动剂T0901317相当。此外,基于Biacore仪器的表面等离子体共振(SPR)技术检测表明,3βHCA对人LXR-α具有较强的结合亲和力(解离常数KD=9.6 μM),显著高于T0901317对人LXR-α的结合亲和力(KD=836 μM)。
为验证LXR-α是否为3βHCA诱导脂肪生成所必需,本研究用LXR-α拮抗剂SR9243处理MPHs。结果显示,SR9243可通过降低生脂相关蛋白表达、减少脂滴数量及降低细胞TG含量,完全阻断3βHCA对脂肪酸合成的促进作用。而IL-22对3βHCA驱动的脂肪生成的抑制效应,与SR9243的作用类似。腺病毒介导的LXR-α敲低实验进一步证实了上述发现:沉默LXR-α可抵消3βHCA诱导的脂肪生成。此外,染色质免疫沉淀(ChIP)实验显示,与flox/flox小鼠相比,HKO小鼠肝脏中LXR-α向Srebf1启动子区域的募集显著增加。综上,这些结果表明LXR-α是3βHCA诱导脂肪生成中不可或缺的关键分子。
图7.3βHCA促进肝细胞内脂肪酸合成
图8.CYP7B1缺失导致的3βHCA蓄积,会加速MPHs及hLOs中的脂肪生成
7.ATF3在HKO小鼠肝脏中抑制Cyp7b1的表达
随后,作者探究了CYP7B1的上游调控因子,发现由Nr0b2编码的非典型核受体——SHP虽已知可影响Cyp7b1表达,但在IL-22过表达或肝脏IL22RA1缺陷后其表达水平未发生改变。因此,研究人员对RNA-seq数据进行全局分析,以筛选调控CYP7B1表达的潜在上游因子。通过分析CD、HFD喂养的IL-22过表达小鼠及HKO小鼠中差异表达的转录因子,鉴定出4个在所有模型中共同重叠的转录因子:Atf3、Srebf1、Nr1i2和Zbtb7c。其中,Atf3和Srebf1在IL-22过表达小鼠与HKO小鼠中呈现相反的基因表达模式;而另外两个转录因子Nr1i2和Zbtb7c则在CD与HFD喂养的IL-22过表达模型间表现出相反的表达趋势。鉴于IL-22在CD和HFD条件下均能减少肝脏TG蓄积,研究者推测目标转录因子的表达应呈现相似趋势。已有研究表明ATF3与肝脂肪变性相关。本研究中,Western blots分析显示,IL-22可降低HFD喂养小鼠的ATF3表达,同时上调CYP7B1表达;相反,与flox/flox对照小鼠相比,HFD喂养的HKO小鼠肝脏中ATF3水平升高,而CYP7B1水平降低。荧光素酶报告基因实验证实ATF3可抑制CYP7B1表达。一致地,对包含206例NAFLD患者肝活检样本的公共转录组数据(GSE135251)分析显示,ATF3与CYP7B1的mRNA表达呈显著负相关。这些数据阐明了ATF3在肝脏中如何作为CYP7B1的转录抑制因子发挥作用。
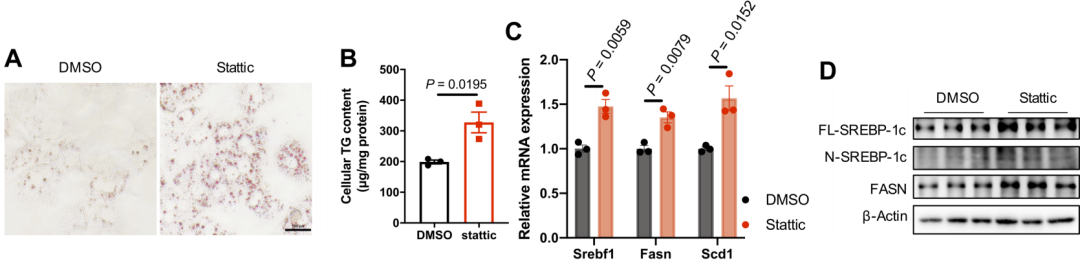
IL-22主要激活Janus激酶/信号转导和转录激活因子(JAK/STAT)通路。因此,通过荧光素酶报告基因实验验证STAT3是否为Atf3的直接上游调控因子,结果显示在HEK293T细胞中STAT3对ATF3表达具有负调控作用。使用STAT3特异性抑制剂Stattic抑制p-STAT3水平后,MPHs中ATF3蛋白水平升高,并导致脂质蓄积增加,这一效应主要通过促进生脂基因的表达实现。值得注意的是,IL-22处理可逆转PA诱导的p-STAT3水平下调,并抑制PA引起的ATF3表达升高。这表明IL-22通过STAT3抑制ATF3,从而调控肝细胞的脂质稳态,在高能量饮食条件下发挥代谢保护作用。
为明确ATF3在HKO小鼠中的作用,研究人员向HKO小鼠来源的原代肝细胞中导入靶向干扰Atf3表达的腺病毒。结果显示,ATF3敲低可上调CYP7B1表达,降低细胞内TG含量,减少脂滴蓄积,其机制主要与这些MPHs中生脂相关蛋白表达下调相关。这提示,在HKO小鼠来源的原代肝细胞中敲低ATF3可恢复CYP7B1表达,从而减少脂质蓄积。用油酸处理HLOs后,ATF3水平及生脂基因表达均显著升高;而IL-22处理可通过抑制HLOs中ATF3的表达,抵消油酸诱导的生脂基因表达升高的效应。
图9.ATF3在HKO小鼠的肝脏中负调控CYP7B1的表达
图10.STAT3是ATF3调控肝细胞脂肪酸合成的上游信号分子
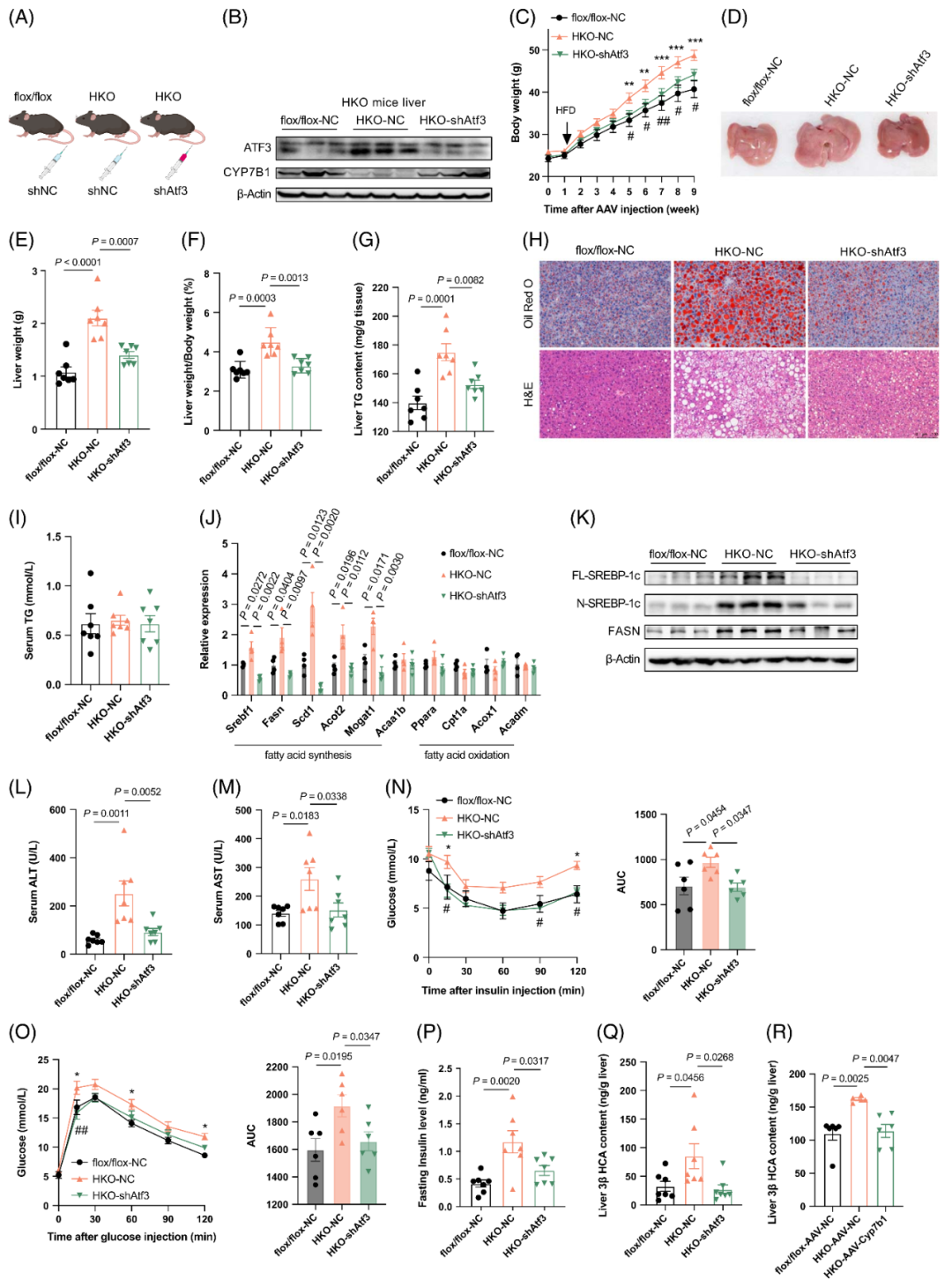
8.HKO小鼠肝脏中ATF3敲低可减轻肝脂肪变性并改善血糖稳态
为评估ATF3在HKO小鼠中的脂质调控作用,研究者采用AAV8(尾静脉注射)靶向敲低小鼠肝脏中的ATF3表达。结果显示,ATF3敲低可上调CYP7B1表达,降低HKO小鼠的体重、肝重及肝重/体重比。在不影响血清TG水平的情况下,ATF3敲低减轻了HKO-shAtf3小鼠的肝脂肪变性。此外,HKO-shAtf3小鼠的血清ALT和AST水平显著降低,表明与HKO-NC小鼠相比,其肝损伤得到缓解。进一步研究发现,ATF3敲低可改善HKO小鼠的胰岛素敏感性和糖耐量,且HKO-shAtf3小鼠的空腹胰岛素水平降低。这些结果表明,干扰ATF3表达可恢复CYP7B1的表达,从而减轻肝脂肪变性并改善血糖稳态。值得注意的是,无论是敲低ATF3还是过表达CYP7B1,均能将HKO小鼠肝脏中的3βHCA含量降至与flox/flox对照小鼠相近的水平。
图11.在HKO小鼠肝脏中沉默ATF3,可减轻肝脂肪变性并改善血糖稳态
总结
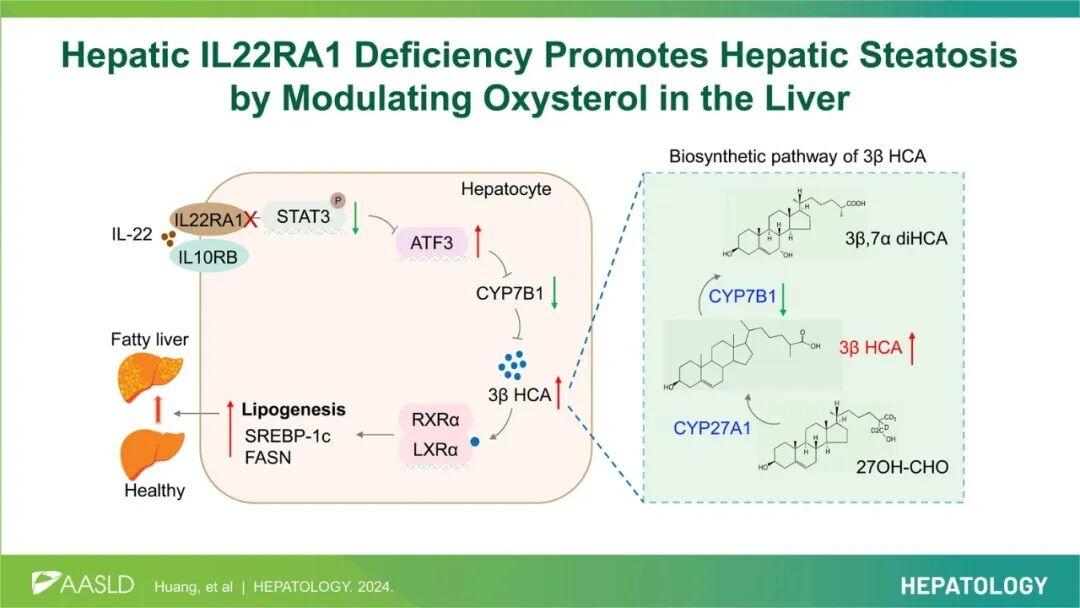
综上,本研究数据表明,肝组织IL22RA1通过ATF3/CYP7B1/3βHCA/LXR-α信号通路调控脂质稳态。本研究揭示了3βHCA的新功能——作为肝细胞脂肪生成的强效激活剂,这一机制可能是肝组织IL22RA1缺陷小鼠肝脏脂质合成异常增加的潜在原因。IL-22可有效抑制3βHCA的生脂效应,表明了IL-22/IL22RA1通路在NAFLD中的治疗潜力。
肝脏IL22RA1缺陷通过3βHCA调控肝脂肪变性的机制模式图
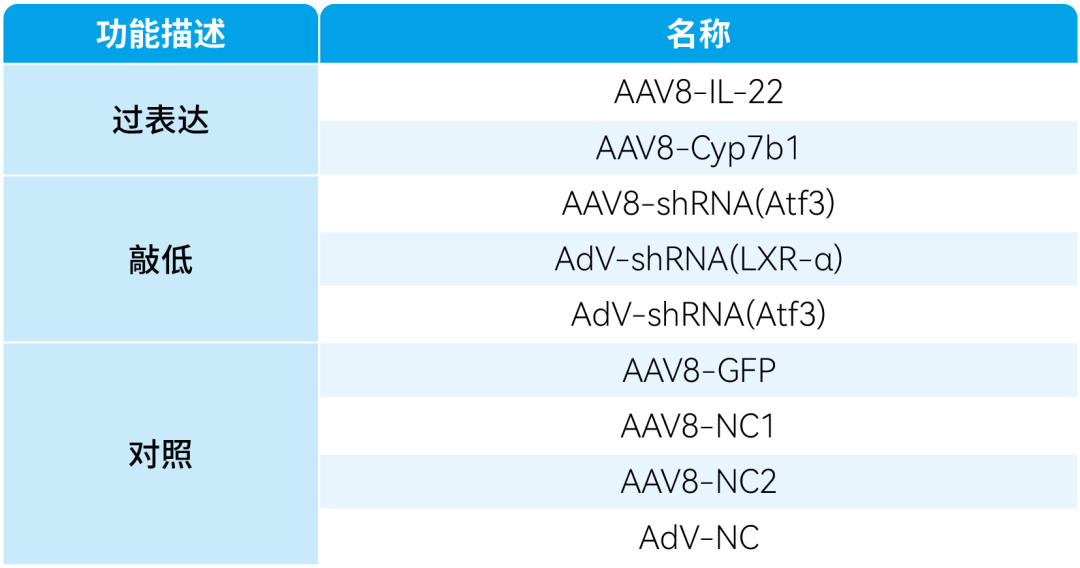
本文使用的病毒产品,列表如下:

了解产品及服务
请扫码添加客服微信:BrainVTA2020

市场:027-65023363 行政/人事:027-62439686 邮箱:marketing@brainvta.com 客服:18140661572(活动咨询、售后反馈等)
销售总监:张经理 18995532642 华东区:陈经理 18013970337 华南区:王经理 13100653525 华中/西区:杨经理 18186518905 华北区:张经理 18893721749
地址:中国武汉东湖高新区光谷七路128号中科开物产业园1号楼
Copyright © 武汉枢密脑科学技术有限公司. All RIGHTS RESERVED.
鄂ICP备2021009124号 DIGITAL BY VTHINK